What is Krabbe Disease?
Krabbe disease, also known as “Globoid Cell Leukodystrophy,” belongs to the group of lysosomal storage disorders (LSDs), specifically the sphingolipidoses. It is a rare, autosomal recessive disorder characterized by the progressive loss of myelin in the nervous system. Myelin, a fatty sheath surrounding nerve cells, is essential for the rapid transmission of nerve signals. In Krabbe disease, abnormal multinucleated cells called “globoid cells” accumulate in the brain, contributing to myelin destruction and severe neurological dysfunction.
It has four clinical subtypes:
- Type 1 Krabbe disease – Infantile
- Type 2 Krabbe disease – Late infantile
- Type 3 Krabbe disease – Juvenile
- Type 4 Krabbe disease – Adult.
What causes Krabbe’s Disease?
Krabbe disease is caused by mutations in the GALC gene, which lead to a deficiency of the enzyme galactosylceramidase. This enzyme is responsible for breaking down galactolipids, including galactosylceramide and psychosine. Without sufficient enzyme activity, these lipids accumulate to toxic levels, damaging oligodendrocytes—the cells responsible for producing myelin.
Excess galactosylceramide leads to the formation of globoid cells, while psychosine accumulation becomes highly toxic, resulting in widespread demyelination in the nervous system. The loss of myelin disrupts nerve signal transmission in the brain and throughout the body, leading to the severe neurological symptoms associated with Krabbe disease.”
Who is at risk of developing Krabbe’s Disease?
Krabbe’s disease affects different populations at different rates. In Europe, it occurs in approximately 1 in 100,000 live births.1 Jain M, De Jesus O. Krabbe Disease. [Updated 2023 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK56231 Since it is an autosomal recessive genetic disorder, a child must inherit two copies of the mutated GALC gene—one from each parent—to develop the disease. Parents who each carry one mutated gene are typically asymptomatic carriers.
The prevalence of Krabbe disease is higher in populations where consanguineous marriages (cousin marriages) are common, as this increases the likelihood of inheriting two mutated copies of the gene.
How is Krabbe’s Disease diagnosed?
Laboratory Testing:
Doctors look for symptoms to diagnose Krabbe’s disease. If they suspect the condition, they send a blood sample or a skin tissue biopsy to a laboratory for testing. The activity of the GALC enzyme in the samples will be measured in the lab. Low GALC activity levels in children may indicate Krabbe disease.
Imaging Studies:
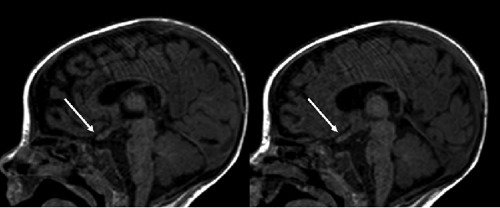
Magnetic Resonance Imaging (MRI)
Magnetic fields are used in this non-invasive procedure to allow the doctor to examine the brain for abnormalities. Krabbe disease causes diffuse demyelination of nerves in children. It can also show optic nerve enlargement in early infantile Krabbe’s.2 Hussain SA, Zimmerman HH, Abdul-Rahman OA, Hussaini SM, Parker CC, Khan M. Optic nerve enlargement in Krabbe disease: a pathophysiologic and clinical perspective. J Child Neurol. 2011 May;26(5):642-4. doi: 10.1177/0883073810387929. Epub 2011 Jan 31. PMID: 21285037.
Computed Tomography (CT)
A CT scan is a type of diagnostic imaging procedure that uses X-rays and computer technology to create detailed images of the internal structures of the body. Krabbes in its early stages will show symmetrical hyperdense areas in the brain. This is due to demyelination. As the condition worsens, hypodense areas appear as a result of atrophy. Hence, Cerebral and cerebellar atrophy appear later in the disease’s progression.
Electromyography & Nerve Conduction
Nerve conduction studies assess the speed at which electrical impulses travel through the nervous system, providing important diagnostic information. Conduction velocity in the motor and sensory nerves slows significantly in infantile Krabbe disease. Around 20% of patients with late-onset Krabbe disease have slowed nerve conduction velocity.3Greco MR, Lopez MA, Beltran-Quintero ML, Tuc Bengur E, Poe MD, Escolar ML. Infantile Krabbe disease (0-12 months), progression, and recommended endpoints for clinical trials. Ann Clin Transl Neurol. 2024 Dec;11(12):3064-3080. doi: 10.1002/acn3.52114. Epub 2024 Nov 5. Erratum in: Ann Clin Transl Neurol. 2025 Feb;12(2):455. doi: 10.1002/acn3.52275. PMID: 39499628; PMCID: PMC11651195.
Genetic Testing:
Genetic testing can help identify the congenital defect that causes Krabbe disease. It can detect at-risk pregnancies, disease carriers, and assist in predicting the genotype’s phenotype. A patient’s blood sample will be required for this.
How is Screening for Krabbe’s Disease done?
Newborn screening for Krabbe disease follows a three-step process:
- Tandem mass spectrometry (TMS) analyzes enzyme activity in newborn blood spots.
- DNA testing is performed if the enzyme activity is low to check for GALC mutations.
- If both tests indicate Krabbe disease, galactocerebrosidase enzyme activity is measured for confirmation.4Lantos JD. Dangerous and expensive screening and treatment for rare childhood diseases: the case of Krabbe disease. Dev Disabil Res Rev. 2011;17(1):15-8. doi: 10.1002/ddrr.133. PMID: 22447750; PMCID: PMC4014301.
Prenatal Diagnosis
Krabbe disease can also be diagnosed before birth using amniocentesis (performed between 15-20 weeks of pregnancy). This test analyzes amniotic fluid for genetic abnormalities.
Symptoms of Krabbe’s Disease
Krabbe’s symptoms are classified as either infantile or late-onset.
Infantile-type symptoms of Krabbe’s disease
- Irritability
- Feeding difficulties
- Hypertonia(muscle stiffness)
- Hyperesthesia (increased sensitivity to stimuli)
- Peripheral neuropathy
- Hyperpyrexia (extremely high fever)
- Psychomotor arrest (loss of motor function development)
- Failure to thrive, vomiting, Gastroesophageal reflux
- Hyperreflexia or hyporeflexia (increased or decreased reflexes)
- Seizures, opisthotonus (severe muscle spasms)
- Rapid neurological deterioration
Late-onset Krabbe’s disease symptoms
- Paresthesias (tingling sensations)
- Decreased muscle strength
- Spasticity
- Ataxia (loss of balance/coordination)
- Paresis (partial paralysis)
- Psychomotor arrest
- Psychomotor deterioration
- Seizures
- Optic atrophy
What are Krabbe’s Disease Treatments?
Krabbe’s disease has no definitive cure. Treatment of Krabbe’s disease necessitates a multidisciplinary approach that includes neurologists, ophthalmologists, geneticists, physiotherapists, and others. The treatment is also most beneficial to patients who are asymptomatic or have only minor symptoms.
Cord Blood Stem Cell Transfusion:
Cord blood stem cells from an unrelated donor are infused into the patient to replace defective microglial cells with ones that produce functional GALC enzyme. This therapy is most effective when performed before symptoms appear and can significantly slow disease progression.5Escolar ML, Poe MD, Martin HR, Kurtzberg J. A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics. 2006 Sep;118(3):e879-89. doi: 10.1542/peds.2006-0747. Epub 2006 Aug 21. PMID: 16923928.
Hematopoietic Stem Cell Transplant (HSCT):
Healthy hematopoietic cells from a donor are transplanted into the patient. This procedure helps to populate the brain with microglia that function normally and have high GALC enzyme activity. HSCT is most effective before symptoms appear and can slow or stop the disease’s progression.6Langan TJ, Barcykowski AL, Dare J, Pannullo EC, Muscarella L, Carter RL. Evidence for improved survival in postsymptomatic stem cell-transplanted patients with Krabbe’s disease. J Neurosci Res. 2016 Nov;94(11):1189-94. doi: 10.1002/jnr.23787. PMID: 27638603; PMCID: PMC5484586. This treatment is also costly, risky, and of uncertain efficacy.7Lantos JD. Dangerous and expensive screening and treatment for rare childhood diseases: the case of Krabbe disease. Dev Disabil Res Rev. 2011;17(1):15-8. doi: 10.1002/ddrr.133. PMID: 22447750; PMCID: PMC4014301.
Supportive Treatment
To manage their symptoms, patients can receive supportive care. Among the possible supportive measures are:
- Physiotherapy to help with mobility
- Occupational therapy is especially beneficial for older children.
- Swallowing impairment leads to tubes being used to ensure proper nutrition.
- Spasm-relaxants and Anticonvulsants, to treat seizures
Life Expectancy of Krabbe’s Disease
Krabbe disease is a progressive and fatal disorder, with life expectancy varying by disease subtype.
- Infantile Krabbe Disease (Type 1): The median lifespan is 13 to 24 months, with most children not surviving beyond age two. Early hematopoietic stem cell transplantation (HSCT) has shown improved survival if performed before symptoms appear.8Krivit W, Shapiro EG, Peters C, Wagner JE, Cornu G, Kurtzberg J, Wenger DA, Kolodny EH, Vanier MT, Loes DJ, Dusenbery K, Lockman LA. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N Engl J Med. 1998 Apr 16;338(16):1119-26.
- Late-Infantile Krabbe Disease (Type 2): Life expectancy varies, but most patients die within 2 to 7 years of symptom onset.
- Juvenile-Onset Krabbe Disease (Type 3): The disease progresses at a moderate rate, with survival ranging from several years to early adulthood, depending on severity and treatment.
- Adult-Onset Krabbe Disease (Type 4): This form progresses more slowly. While some patients survive for decades, others experience significant neurological decline and may live only 5 to 7 years after symptom onset.
Prevention of Krabbe’s Disease
Provide genetic counseling for at-risk couples to explain reproductive options. Prenatal diagnosis, if feasible and desired, can be beneficial in future pregnancies by providing reassurance in the case of an unaffected fetus or by allowing an informed exploration of options, such as termination of pregnancy or, potentially, early stem cell therapy, in the case of an affected fetus. To completely prevent the disease carriers of Krabbe’s must not have children. In consanguineous marriages, where both parents carry a disease, the disease can affect their child with a 25% probability. Typically, neither parent will exhibit any symptoms of the condition. People with a Krabbe family history should seek genetic counseling.
Conclusion
Krabbe’s disease, a leukodystrophy, results from a loss of myelin in the nervous system. Mutations in the GALC gene primarily cause the disease, leading to a deficiency of the galactosylceramidase enzyme and the buildup of harmful substances in the nervous system. Krabbe’s disease encompasses several subtypes, with the most common one manifesting before the age of one. Late-onset forms, in contrast, may emerge during childhood, adolescence, or adulthood, exhibiting a range of symptoms.
Doctors use a blood test to assess GALC enzyme levels and detect potential cases of Krabbe disease early on. MRI, CT, electromyography, nerve conduction studies, and genetic testing are all used to help confirm the diagnosis. Unfortunately, there is no cure for Krabbe disease. Treatment of Krabbe’s disease focuses primarily on symptom management and supportive care.
The disease can cause severe complications such as deafness, vision loss, cognitive decline, and respiratory failure, which can lead to death. The disease’s form determines life expectancy, with infantile cases rarely surviving beyond two years and late-onset cases surviving up to 5 to 7 years after symptom onset.
Preventing Krabbe’s disease requires genetic counseling for GALC gene carriers, particularly in consanguineous marriages. Knowing one’s disease carrier status can help people make informed family planning decisions to reduce the risk of passing the disease on to their children.
Refrences
- 1Jain M, De Jesus O. Krabbe Disease. [Updated 2023 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK56231
- 2Hussain SA, Zimmerman HH, Abdul-Rahman OA, Hussaini SM, Parker CC, Khan M. Optic nerve enlargement in Krabbe disease: a pathophysiologic and clinical perspective. J Child Neurol. 2011 May;26(5):642-4. doi: 10.1177/0883073810387929. Epub 2011 Jan 31. PMID: 21285037.
- 3Greco MR, Lopez MA, Beltran-Quintero ML, Tuc Bengur E, Poe MD, Escolar ML. Infantile Krabbe disease (0-12 months), progression, and recommended endpoints for clinical trials. Ann Clin Transl Neurol. 2024 Dec;11(12):3064-3080. doi: 10.1002/acn3.52114. Epub 2024 Nov 5. Erratum in: Ann Clin Transl Neurol. 2025 Feb;12(2):455. doi: 10.1002/acn3.52275. PMID: 39499628; PMCID: PMC11651195.
- 4Lantos JD. Dangerous and expensive screening and treatment for rare childhood diseases: the case of Krabbe disease. Dev Disabil Res Rev. 2011;17(1):15-8. doi: 10.1002/ddrr.133. PMID: 22447750; PMCID: PMC4014301.
- 5Escolar ML, Poe MD, Martin HR, Kurtzberg J. A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics. 2006 Sep;118(3):e879-89. doi: 10.1542/peds.2006-0747. Epub 2006 Aug 21. PMID: 16923928.
- 6Langan TJ, Barcykowski AL, Dare J, Pannullo EC, Muscarella L, Carter RL. Evidence for improved survival in postsymptomatic stem cell-transplanted patients with Krabbe’s disease. J Neurosci Res. 2016 Nov;94(11):1189-94. doi: 10.1002/jnr.23787. PMID: 27638603; PMCID: PMC5484586.
- 7Lantos JD. Dangerous and expensive screening and treatment for rare childhood diseases: the case of Krabbe disease. Dev Disabil Res Rev. 2011;17(1):15-8. doi: 10.1002/ddrr.133. PMID: 22447750; PMCID: PMC4014301.
- 8Krivit W, Shapiro EG, Peters C, Wagner JE, Cornu G, Kurtzberg J, Wenger DA, Kolodny EH, Vanier MT, Loes DJ, Dusenbery K, Lockman LA. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N Engl J Med. 1998 Apr 16;338(16):1119-26.
