Niemann-Pick Disease is a lipid-storage disorder marked by the excessive accumulation of lipids, including waxes, oils, and cholesterol, in various organs such as the brain, spleen, liver, lungs, and bone marrow.
This condition is characterized by a deficiency in the lysosomal enzyme acid sphingomyelinase, leading to the buildup of sphingomyelin in cells and tissues. Inadequate or malfunctioning enzymes hinder the breakdown of lipids into smaller components, depriving the normal energy supply. This disease can affect critical organs, including the brain, nerves, liver, spleen, bone marrow, and, in severe cases, the lungs.1Bajwa, H., & Azhar, W. (2023). Niemann-Pick Disease. In StatPearls. StatPearls Publishing The progressive loss of nerve, brain, and other organ functions is associated with symptoms of the niemann-pick disease. Although it can happen to anyone, Niemann-Pick primarily affects youngsters. The illness is occasionally lethal and has no recognized treatment.
Types of Niemann-Pick Disease
There are three types of disease.
Type A:
Type A, the most severe variant, typically manifests in early infancy and predominantly affects Jewish families. By six months of age, children with this type exhibit profound brain damage and weakness, with a lifespan rarely extending beyond 18 months.2Hu, J., Maegawa, G. H., Zhan, X., Gao, X., Wang, Y., Xu, F., … & Zhang, H. (2021). Clinical, biochemical, and genotype‐phenotype correlations of 118 patients with Niemann‐Pick disease Types A/B. Human Mutation, 42(5), 614-625.
Type B:
Type B of Niemann-Pick-disease, also known as Juvenile onset, typically manifests as peripheral neuropathy (nerve damage and disturbed signaling), ataxia, and other symptoms in the preteen years. Typically, the brain is unaffected. Sphingomyelin, a fatty molecule found in every body cell, builds up in dangerous proportions in types A and B due to insufficient enzyme activity. Children with type B may live relatively long lives, but because of lung damage, they may need extra oxygen.3 Hu, J., Maegawa, G. H., Zhan, X., Gao, X., Wang, Y., Xu, F., … & Zhang, H. (2021). Clinical, biochemical, and genotype‐phenotype correlations of 118 patients with Niemann‐Pick disease Types A/B. Human Mutation, 42(5), 614-625
Type C:
Type C may present in early childhood or develop during adolescence or adulthood. It is attributed to a deficiency of NPC1 or NPC2 proteins. Neurological complications such as extensive brain damage may lead to challenges in basic functions like looking up and down, walking, and swallowing, alongside a gradual decline in vision and hearing. The severity of individual cases can vary, with some individuals succumbing to the condition during childhood while others manage to live into adulthood, depending on their specific circumstances.4Las Heras, M., Szenfeld, B., Ballout, R. A., Buratti, E., Zanlungo, S., Dardis, A., & Klein, A. D. (2023). Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. NPJ Genomic Medicine, 8(1), 21.
Causes of Niemann-Pick Disease
It is an autosomal recessive (AR) disease, which implies that both parents must have the condition or be gene carriers for their offspring to contract it. Missense mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene lead to NPD types A and B. In SMPD1, more than 180 mutations have been found. Mutations in the NPC1 and NPC2 genes found on chromosomes 18 and 14 are the root cause of NPD type C. These genes’ abnormalities result in incorrect or defective protein synthesis, which hinders the passage of lipids out of the cells and causes their buildup. This causes the characteristic appearance of ‘Foam cells‘ in NPD.5Wheeler, S., & Sillence, D. J. (2020). Niemann-Pick type C disease: cellular pathology and pharmacotherapy. Journal of Neurochemistry, 153(6), 674–692. https://doi.org/10.1111/jnc.14895
Although carriers of an AR disease do not exhibit the disease’s signs and symptoms, they can nonetheless convey the faulty gene to the next generation. Therefore, each pregnancy has a 25% chance of giving birth to a kid who has the condition if both parents are carriers.
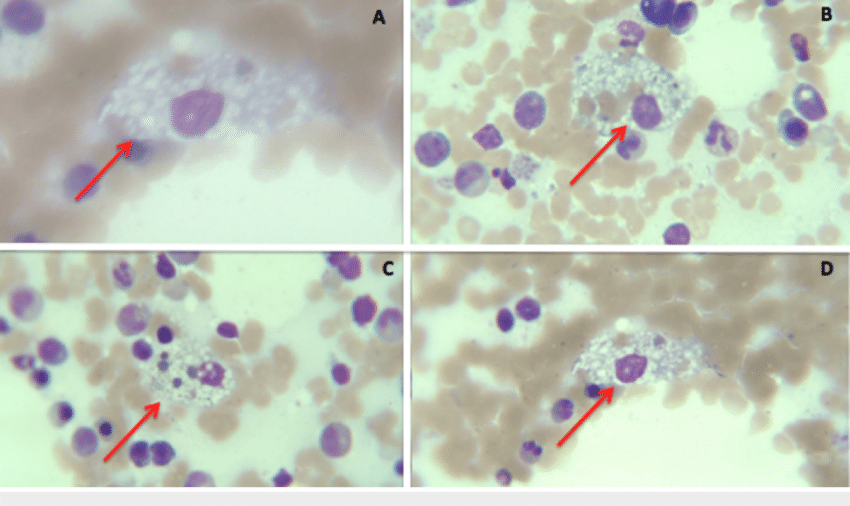
(Distributed under license CC BY)
Symptoms of Niemann-Pick Disease
Depending on the type and severity, people with the Niemann-pick disease may encounter certain symptoms. Within the first few months of life, some newborns with type A will begin to exhibit signs and symptoms.6 Matencio, A., Navarro-Orcajada, S., González-Ramón, A., García-Carmona, F., & López-Nicolás, J. M. (2020). Recent advances in the treatment of Niemann pick disease type C: A mini-review. International Journal of Pharmaceutics, 584, 119440. Type B patients are more likely to live to adulthood, even if they may not exhibit symptoms for years. Those with type C may not exhibit symptoms until they are adults. Some signs could be:
- Liver and spleen enlargement (hepatosplenomegaly)
- Ataxia (A problem in controlling movement)
- Vertical Supranuclear Gaze palsy: irregular eye movements
- Muscular atrophy (hypotonia)
- Presence of common respiratory diseases
- Challenges in speaking
- Difficulties with eating and swallowing
- Diminished mental capacity
- Occurrence of seizures
Other neurological manifestations associated with NPD may involve drooling and cataplexy. Cataplexy, marked by a sudden loss of muscle tone and strength, can result in abrupt head drops, a weak, rubbery feeling in the legs, or, in severe instances, complete collapse.
Diagnosis of Niemann-Pick Disease
A thorough physical examination is the first step in diagnosing Niemann-Pick disease and may reveal an early warning indication such as an enlarged liver or spleen. The doctor will also review the symptoms and the family’s health history while taking a thorough medical history. This disease is an uncommon condition whose symptoms can be mistaken for those of other illnesses7Berry-Kravis, E. (2021, April). Niemann-Pick disease, type C: diagnosis, management and disease-targeted therapies in development. In Seminars in Pediatric Neurology (Vol. 37, p. 100879). WB Saunders. Various Niemann-Pick disease subtypes require different diagnostic approaches.
- Type A or B: The amount of sphingomyelinase in white blood cells is measured by professionals using a blood or skin sample (biopsy) to confirm the diagnosis.
- Type C: To evaluate how the cells move and store cholesterol, experts take a tiny skin sample to test for Niemann-Pick disease.
Filipin Staining & Cholesterol Esterification Test:
Traditionally, confirming a Niemann-Pick Disease (NPD) diagnosis involved Filipin staining of the individual’s skin cells (fibroblasts). This process assessed the level of cholesterol accumulation. Additionally, the Cholesterol Esterification Test was employed to gauge the cells’ ability to modify cholesterol. While these methods were once standard diagnostic procedures, advancements in diagnostic technology have introduced more contemporary approaches.
Blood-Based Biomarker Testing:
In recent years, the diagnostic landscape for NPD has shifted towards blood-based testing for biomarkers. This modern approach involves analyzing blood samples for specific biomarkers such as oxysterols, lysosphingolipids, and bile acid metabolites. This method offers a more convenient and efficient diagnosis method than traditional staining techniques.
Molecular Gene Sequencing of NPC1 & NPC2:
A significant leap in NPD diagnosis has been the adoption of molecular gene sequencing. This advanced method entails sequencing genes NPC1 and NPC2 to identify genetic abnormalities associated with NPD. Molecular gene sequencing has become a cornerstone in diagnosing NPD (Type C), surpassing the limitations of traditional staining and cholesterol esterification tests. This approach enhances accuracy and streamlines the diagnostic process for individuals suspected of having Niemann-Pick Disease.8National Organization for Rare Disorders. (n.d.). Niemann-Pick Disease Type C. Retrieved from https://rarediseases.org/rare-diseases/niemann-pick-disease-type-c/#disease-overview-main
Additionally, the following examinations may also help in the diagnosis:
Magnetic Resonance Imaging (MRI):
A brain MRI may show a loss of brain cells. However, an MRI might be normal in the early stages of this disease, and symptoms frequently appear before brain cell death.
Eye Examination:
Eye exams can reveal symptoms of Niemann-Pick disease, such as problems with eye movement.
In infants affected by the condition, noticeable cherry red spots typically emerge within the initial 12 months of life, and the entire retina takes on an ‘opaque’ appearance. Intracellular lipid accumulation has been observed in various eye components, including retinal neurons, amacrine cells, retinal pigment epithelial cells, and receptors.
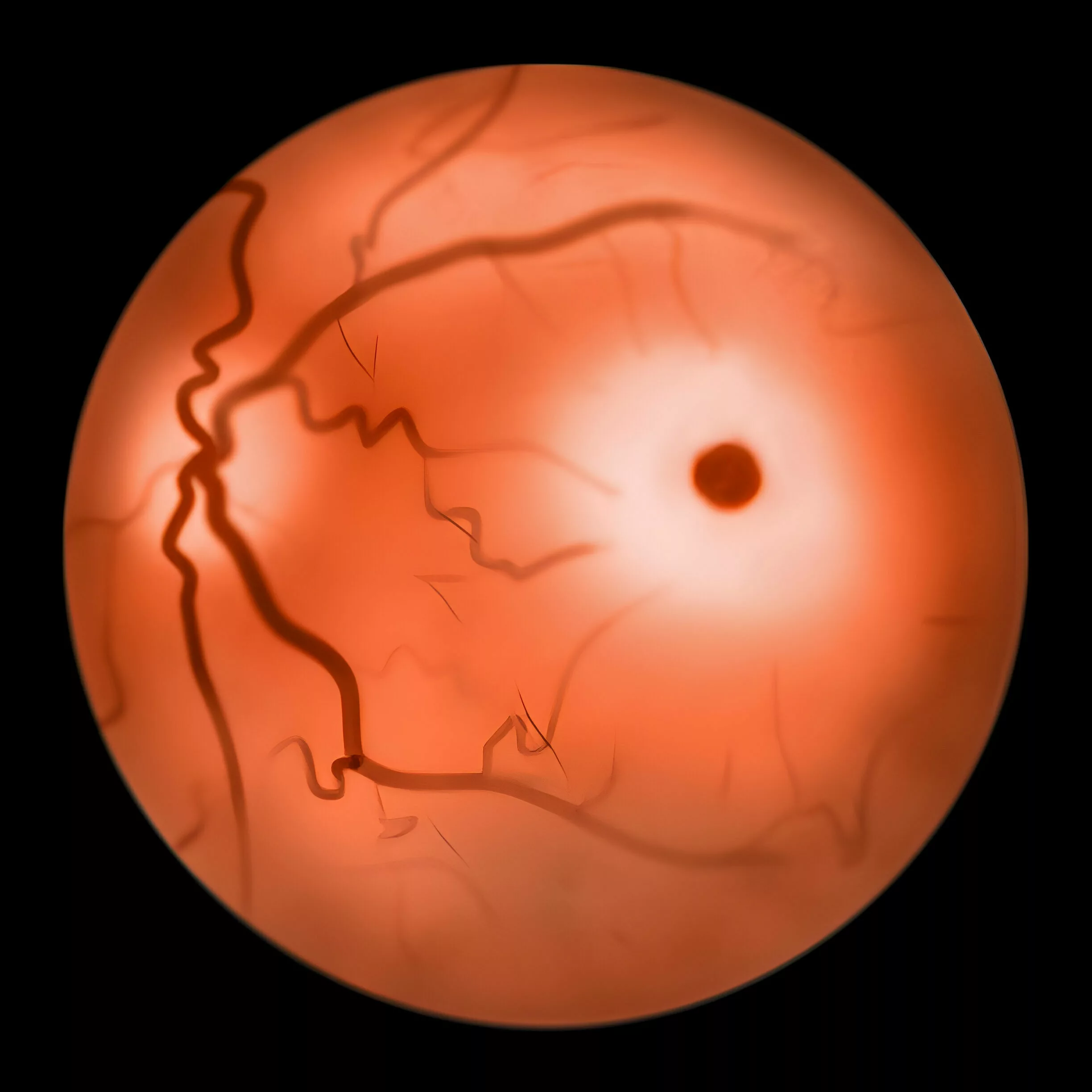
Prenatal Examination:
Ultrasound can reveal the enlarged liver and spleen caused by type C. Additionally, confirming a Niemann-Pick diagnosis may involve utilizing chorionic villus collection or amniocentesis.
Differential Diagnosis of Niemann-Pick Disease
Keep other lysosomal storage disorders in the differential diagnosis, including Gaucher’s disease, Tay-Sachs disease, and Metachromatic leukodystrophy. Hepatosplenomegaly and cytopenia are also symptoms of Gaucher disease, but bone discomfort and lesions are more noticeable. Because glucocerebrosidase is lacking in Gaucher disease, glucocerebroside accumulates inside cells as opposed to sphingomyelin, which is present in this disease.
On the other hand, Tay-Sachs disease is characterized by neurodegeneration, developmental delay, and a cherry-red patch on the macula. Hexosaminidase A deficiency is the culprit in this case, which results in an accumulation of GM2 gangliosides. However, there is an absence of hepatosplenomegaly.
Ataxia and other neurological symptoms are possible manifestations of metachromatic leukodystrophy, resulting in central and peripheral demyelination.9Bajwa, H., & Azhar, W. (2023). Niemann-pick disease. In StatPearls [Internet]. StatPearls Publishing.

Treatment of Niemann-Pick Disease
The available treatments will depend on the type of Niemann-Pick disease.
Type A
For Type A, which often results in death within the first few years of life, there are currently no treatments available. However, symptoms are treated.
Type B
However, for Type B, enzyme replacement therapy, bone marrow transplants, and gene therapy, which involves replacing unhealthy genes with good ones, are all ways that doctors treat NPB. Recombinant acid sphingomyelinase may be used as an enzyme replacement therapy in some cases of Niemann-Pick disease. All these treatments can only help people with NPB live longer, but none of them can cure it.
Type C
For Type C, doctors use a variety of physical therapies and medications. Moreover, migulstat, an enzyme inhibitor that people frequently take to stop their bodies from creating fatty compounds like cholesterol, has been approved for treating progressive neurological manifestations in adult patients and pediatric patients with NPC (Niemann-Pick disease Type C). This therapy lessens fat accumulation, which lessens the consequences of type C.10Patterson, M. C., Mengel, E., Vanier, M. T., Moneuse, P., Rosenberg, D., & Pineda, M. (2020). Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: a final report of the NPC Registry. Orphanet journal of rare diseases, 15, 1-10.
Life Expectancy of Niemann-Pick Disease
The prognosis and life expectancy differ according to how each kind of Niemann-Pick disease presents itself. Even though everyone with the condition has it from birth, the symptoms sometimes don’t show up until much later in life. When symptoms first occur in childhood or adolescence, the life expectancy is frequently less than a few years. However, life expectancy increases to maturity if they appear after the first few years.
- In Type A patients, due to the severity of the symptoms, most children with NPA do not live through their first few years.
- However, for type B, preadolescence is when NPB, also known as juvenile-onset Niemann-Pick disease, typically manifests. Although it exhibits some of the same symptoms as NPA, they are typically milder. Individuals with NPB frequently survive into maturity.
- Similarly, for type C, the prognosis for affected individuals depends on when issues start.
Complications of Niemann-Pick Disease
It is a degenerative condition, and problems frequently arise over time.
- The liver’s early involvement may progress to fulminant hepatic failure.
- Lung deterioration can cause respiratory failure.
- Progressive neurodegeneration can cause dementia, convulsions, and schizophrenia-like psychosis.
- Internal or external bleeding might occur because of severe thrombocytopenia.
- Valvular heart disease and coronary artery disease
- Deformed bones can result in coxa vara (a deformity of the hip whereby the angle between the head and the shaft of the femur is reduced to less than 120 degrees), larger bone marrow cavities, or thinner cortical bone.
In summary, Niemann-Pick Disease is a complex set of hereditary metabolic disorders with genetic origins. Advances in diagnostic methods offer hope for improved precision in identification and management. As we make progress, it’s crucial to keep spreading awareness and building a supportive community. These aspects play a vital role in caring for individuals dealing with Niemann-Pick Disease.
Refrences
- 1Bajwa, H., & Azhar, W. (2023). Niemann-Pick Disease. In StatPearls. StatPearls Publishing
- 2Hu, J., Maegawa, G. H., Zhan, X., Gao, X., Wang, Y., Xu, F., … & Zhang, H. (2021). Clinical, biochemical, and genotype‐phenotype correlations of 118 patients with Niemann‐Pick disease Types A/B. Human Mutation, 42(5), 614-625.
- 3Hu, J., Maegawa, G. H., Zhan, X., Gao, X., Wang, Y., Xu, F., … & Zhang, H. (2021). Clinical, biochemical, and genotype‐phenotype correlations of 118 patients with Niemann‐Pick disease Types A/B. Human Mutation, 42(5), 614-625
- 4Las Heras, M., Szenfeld, B., Ballout, R. A., Buratti, E., Zanlungo, S., Dardis, A., & Klein, A. D. (2023). Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. NPJ Genomic Medicine, 8(1), 21.
- 5Wheeler, S., & Sillence, D. J. (2020). Niemann-Pick type C disease: cellular pathology and pharmacotherapy. Journal of Neurochemistry, 153(6), 674–692. https://doi.org/10.1111/jnc.14895
- 6Matencio, A., Navarro-Orcajada, S., González-Ramón, A., García-Carmona, F., & López-Nicolás, J. M. (2020). Recent advances in the treatment of Niemann pick disease type C: A mini-review. International Journal of Pharmaceutics, 584, 119440.
- 7Berry-Kravis, E. (2021, April). Niemann-Pick disease, type C: diagnosis, management and disease-targeted therapies in development. In Seminars in Pediatric Neurology (Vol. 37, p. 100879). WB Saunders.
- 8National Organization for Rare Disorders. (n.d.). Niemann-Pick Disease Type C. Retrieved from https://rarediseases.org/rare-diseases/niemann-pick-disease-type-c/#disease-overview-main
- 9Bajwa, H., & Azhar, W. (2023). Niemann-pick disease. In StatPearls [Internet]. StatPearls Publishing.
- 10Patterson, M. C., Mengel, E., Vanier, M. T., Moneuse, P., Rosenberg, D., & Pineda, M. (2020). Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: a final report of the NPC Registry. Orphanet journal of rare diseases, 15, 1-10.
