Carpenter Syndrome, also known as acrocephalopolysyndactylyl type Ⅱ, is a rare genetic disorder that causes multiple deformities in infants. It is primarily caused by mutations in the RAB23 gene, which affect normal development. One of the hallmark features is craniosynostosis, the premature fusion of skull bones, leading to an abnormally shaped head, distinctive facial features, and sometimes hearing and vision problems. Deformities of fingers (hands/feet), skeletal and spinal problems, along with heart defects, are also frequently seen in cases of Carpenter syndrome. According to the National Organization for Rare Disorders, only about 70 cases have been reported of this extremely rare inherited condition.1National Organization for Rare Disorders (NORD). (2021). Carpenter syndrome. RareDiseases.org. https://rarediseases.org/rare-diseases/carpenter-syndrome/ The severity of the disease varies between individuals. Treatment involves symptomatically managing the complications. It generally requires a multi-disciplinary team of professionals for management.
Carpenter Syndrome Symptoms
The premature closure of the skull bones leads to manifestations in different parts of the body (and not only the head and neck region). The rigid cephalic structure hinders the growth of the baby’s brain. This eventually leads to havoc throughout the body. However, the intensity of the symptoms can vary from mild to severe disabilities (especially intellectual disability). The most common symptoms of carpenter syndrome include:
Facial & Skull Asymmetries
Carpenter syndrome patients have a typical cloverleaf-shaped skull. The head is short and broad, i.e., a condition called brachycephaly. Severe brachycephaly can be seen in cases with the fusion of multiple sutures, including bilateral coronal sutures, lambdoid, and sagittal sutures.2Bouaré, F., Noureldine, M. H. A., Hajhouji, F., Ghannane, H., Jallo, G. I., & Ait Benali, S. (2022). Complex craniosynostosis in the context of Carpenter’s syndrome. Child’s Nervous System, 38(4), 831-835. Premature craniosynostosis may also lead to a pointy/tower or conical head skull known as acrocephaly.
The uneven fusing of the skull bones leads to an asymmetric cranium. The asymmetry follows in the face as well, and common facial malformations include:
- Flat nose (nasal bridge)
- Widely spaced eyes (eye sockets appear shallow)
- Low-set ears
- Downward slanting eyelid folds (palpebral fissures)
- Underdeveloped jaws (upper and lower)3Asadi, S., Amjadi, H., & Jamali, M. (2019). The role of genetics mutations in genes rab23 & megf8 in Carpenter syndrome. Journal of Bioences, 3(5), 1-8.
Dental deformities like primary hypodontia (small-sized milk teeth) may accompany the short-sized upper and lower jaws.
Digit (Fingers) Abnormalities
Abnormally formed fingers are salient features of the genetic disease. Clinicians have seen multiple types of digital defects, including:
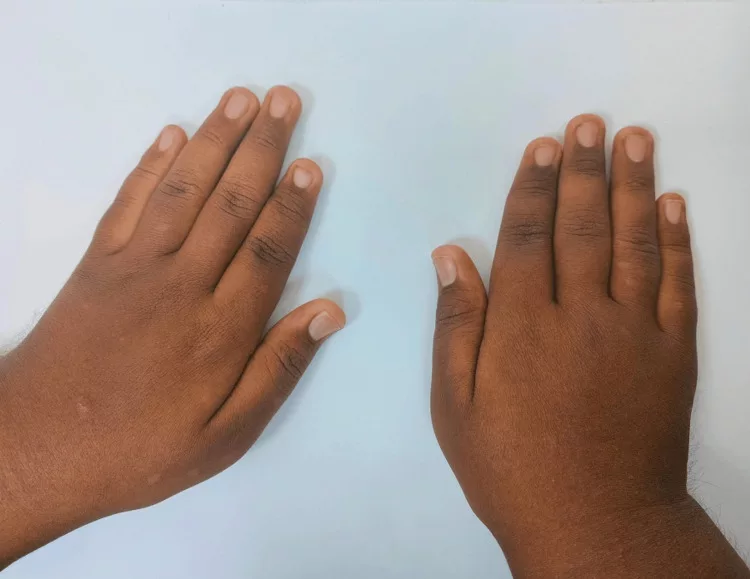
- Cutaneous syndactyly: Fusion of the skin of 2 (or more) fingers. It can be present in fingers and toes. Cutaneous syndactyly is commonly seen between the third and fourth fingers.
- Brachydactyly: Short fingers or toes are present in cases of the syndrome.
- Polydactyly: Patients often possess extra fingers. The most common site for the extra finger is next to the big or second toe. An additional digit may also grow near the pinky finger. 4Batta, A. (2019). Carpenter Syndrome—A Genetic Disease.
Intellectual Disabilities
Individuals may encounter mild to severe mental disability depending on the timing of the bone fusion. The fusion of the bones prevents the normal growth (and expansion) of the brain which may lead to intellectual disabilities and mental retardation. However, normal intelligence is seen in many instances.
Spinal & Skeletal Deformities
Multiple issues may arise in the skeletal and spinal structures of the body. Patients suffer from coxa valga (a disorder in which the angle between the head and neck of the femur and the shaft is increased). This usually accompanies genu valgum (a condition in which the knees are angled inwards). In addition to the knee and hip issues, patients also develop a rounded and curved upper back,i.e., kyphoscoliosis.
Heart Abnormalities
Infants often experience multiple congenital heart defects that often require early medical attention. There have been cases of a degree Ⅰ atrioventricular block (a disorder that hinders the conduction of electrical signals from the atria of the heart to the ventricles) and enlargement of the right atrium (chamber of the heart). Defects of the cardiac vessels, like subaortic ventricular septal defect (hole in the heart’s wall that separates the heart’s chambers) and infundibular/valvular pulmonary stenosis (narrowing of the pulmonary valve that hinders blood flow), may also be present.5Güvenç, O., Çimen, D., Arslan, D., & Güler, İ. (2016). Coexistence of Carpenter syndrome and double outlet right ventricle.
Turk Kardiyol Dern Ars ,
45 (5), 454-457. There may also be mirror-imaging positioning of the heart, i.e., dextrocardia.
Less Common Issues
- Obesity: Syndromic forms of obesity (like Carpenter and Kleefstra syndrome) have an underlying genetic aspect and mostly accompany issues like developmental delays and congenital malformations.6Carvalho, L. M. L., Jorge, A. A. D. L., Bertola, D. R., Krepischi, A. C. V., & Rosenberg, C. (2024). A comprehensive review of syndromic forms of obesity: genetic etiology, clinical features, and molecular diagnosis. Current Obesity Reports, 13(2), 313-337.
- Genital issues: Male patients suffering from the disorder often suffer from cryptorchidism, i.e., a condition in which testicles do not descend out of the body (into the scrotum). This condition is common to other genetic disorders (like kleefstra syndrome) as well.
- Umbilical hernia: A loop of the small intestine pushes out (herniates) through the belly button (umbilicus). This happens due to a weakening of the abdominal wall.
- Hearing issues: Craniofacial abnormalities can lay the foundation for hearing loss due to conductive or sensorineural hearing loss.
- Vision issues: Visual impairments are sometimes present, and attributed to underdeveloped corneas and optic atrophy.
What Causes Carpenter Syndrome?
It is an inherited genetic disorder, which means the mutations in the genes are transferred from parents to children and passed down between families. Experts are unclear about how these mutations occur in the first place. However, it is an autosomal recessive disorder, which means that a child must possess (inherit) two copies of the mutated genes (one from each parent) for the disease to develop. In most cases, parents do not have the disorder themselves and are mere “carriers” of the disease. According to genetic probability, a child has a 25% risk of Carpenter syndrome if both parents (biological) are carriers of the mutated gene.
Carpenter syndrome arises due to mutations in one or two genes:
- RAB23
- MEGF8
The RAB23 gene is involved in the production of the RAB23 protein, which is responsible for vesicle trafficking (transport of materials) across the cells. This protein regulates developmental pathways (hedgehog signaling pathway), which are important for cell growth and the normal shaping of body parts.7Chau, Y. Y., Liang, H., Tung, W. L., Hor, C. H. H., & Aik, W. S. (2025). Structural basis for Rab23 activation and a loss-of-function mutation in Carpenter syndrome. Journal of Biological Chemistry, 301(1).
The role of the MEGF8 gene is unclear but experts believe that protein may have a role in cell processes like cell adhesion and interaction of proteins with each other. Thus, there may be a contribution of the gene in normal body patterning. Mutations in these genes lead to abnormal development of bodily structures.
Carpenter Syndrome Diagnosis
Physical Examination
Diagnosing genetic syndromes requires multiple tests because of the wide array of presentations. Healthcare providers start with a clinical examination of features like a small skull, webbed (and excess) fingers, facial deformities, etc.
Diagnostic Tests
If suspected, your health provider may order some diagnostic tests. Multiple X-rays and/or CT scans of the involved structures, including the skull, spine, digits, and heart, may be conducted to check for the extent of deformity. Sometimes, an MRI scan may also be required for better visualization of the organs.
An electrocardiogram (ECG) is done to evaluate the performance of the heart. Your doctor may also perform vision and hearing tests to check associated impairments.
Genetic Testing
It is a crucial test to confirm Carpenter syndrome. Molecular genetic testing of genes (RAB23 and MEGF8) helps confirm the diagnosis. The doctor can obtain a DNA sample from your:
- Blood
- Hair
- Skin
Some physicians advise parents to undergo genetic testing during pregnancy. Prenatal genetic testing (preimplantation genetic testing) aids in screening causative familial mutations. Doctors receive a sample from:
- The placenta (chorionic villus sampling)
- The amniotic fluid, which submerges the baby in the womb (amniocentesis)
Prenatal testing is usually paired with a prenatal ultrasound to identify head, heart, and digit abnormalities.
Differential Diagnosis:
Carpenter Syndrome Vs Greig Cephalopolysyndactyly (GCPS)
Both are genetic disorders, but there are subtle differences. While carpenter arises due to mutations in the RAB23 and MEGF8 genes, GCPS develops due to gene mutation/deletion on chromosome 7p14.1. Major issues include polydactyly, mild intellectual issues, and skull deformities. However, unlike Carpenter syndrome, heart problems, severe intellectual issues, and vision issues are not commonly seen.
Carpenter Syndrome Vs Apert Syndrome
Acrocephalopolysyndactylyl type Ⅱ and Apert are identical but Carpenter’s is an autosomal recessive disorder while Apert syndrome is an autosomal dominant disorder. Obesity and polydactyly are commonly seen in Carpenter’s syndrome but less common in Apert.
Carpenter Syndrome Treatment
As it is a genetic disorder, there is no cure for it. However, most cases require a multi-disciplinary team of consultants (dentists, cardiologists, neurologists, occupational therapists, orthopedists, etc.) to manage the wide array of abnormalities. The main aim of the treatment is to reduce disability, boost life quality, and improve the patient’s craniofacial appearance. Addressing heart concerns is important as it can prove to be life-threatening.
Therapies
Physical therapy improves mobility and muscle strength. Occupational therapy helps the patient learn to perform day-to-day activities (like reading and writing) with ease. Speech therapy helps with communication (in cases of intellectual disabilities).
Cranial Surgery
Patients may have to undergo different types of surgical procedures to correct deformities. Cranial vault remodeling (craniosynostosis repair) is done to correct the premature skull bone closure. The vault reshaping allows a reduction in intracranial pressure (pressure of the skull on the brain). The timing of the surgery is a crucial aspect of syndromic craniosynostosis. Clinical studies suggest that if cranial pressure elevation isn’t a concern, remodeling surgery can be done within 6 to 9 months of age.8Utria, A. F., Mundinger, G. S., Bellamy, J. L., Zhou, J., Ghasemzadeh, A., Yang, R., … & Dorafshar, A. H. (2015). The importance of timing in optimizing cranial vault remodeling in syndromic craniosynostosis. Plastic and reconstructive surgery, 135(4), 1077-1084.
Finger Surgery
Reconstructive surgery is done to correct digital abnormalities like syndactyly, polydactyly, etc. Surgeons separate webbed fingers and remove extra digits.
Cardiac Surgery
Cardiac surgeons perform heart surgeries according to the type of defect. Common procedures include the closure of holes in the heart and the correction of other malformations.
Carpenter Syndrome Life Expectancy
The life expectancy for Carpenter syndrome is variable. Most individuals can live a typical life with timely diagnosis and corrective surgeries (at the right time). Individuals with intellectual disability rely on others for basic activities. However, the severity of the disease determines the life expectancy, and a shortened lifespan is seen in cases with severe issues (especially heart complications).
Unfortunately, there is no way to prevent carpenter syndrome.
Final Word
Carpenter syndrome is a rare genetic, inherited disorder that affects the skull, spine, heart, and fingers. Infants suffer from premature closure (fusion) of skull bones, have conjoined (syndactyly) or extra digits (polydactyly), typical craniofacial features (brachycephaly, flat nasal bridge, etc.), cardiac deformities, and intellectual disabilities, etc. Mutations in the RAB23 and MEGF8 genes cause the disease to develop. Diagnosis of Carpenter syndrome requires multiple radiographic tests (X-rays, CT scans, etc.), an ECG (for the heart), and genetic testing. Treatment involves a multi-disciplinary approach to managing the wide array of symptoms, including occupational, speech, and physical therapy. Surgeons perform cranial vault reshaping and digit reconstruction surgeries to correct head/face and finger deformities, respectively. Cases with minor heart defects and lesser intellectual disabilities can live a normal life with the right interventions.
Refrences
- 1National Organization for Rare Disorders (NORD). (2021). Carpenter syndrome. RareDiseases.org. https://rarediseases.org/rare-diseases/carpenter-syndrome/
- 2Bouaré, F., Noureldine, M. H. A., Hajhouji, F., Ghannane, H., Jallo, G. I., & Ait Benali, S. (2022). Complex craniosynostosis in the context of Carpenter’s syndrome. Child’s Nervous System, 38(4), 831-835.
- 3Asadi, S., Amjadi, H., & Jamali, M. (2019). The role of genetics mutations in genes rab23 & megf8 in Carpenter syndrome. Journal of Bioences, 3(5), 1-8.
- 4Batta, A. (2019). Carpenter Syndrome—A Genetic Disease.
- 5Güvenç, O., Çimen, D., Arslan, D., & Güler, İ. (2016). Coexistence of Carpenter syndrome and double outlet right ventricle.
Turk Kardiyol Dern Ars ,
45 (5), 454-457. - 6Carvalho, L. M. L., Jorge, A. A. D. L., Bertola, D. R., Krepischi, A. C. V., & Rosenberg, C. (2024). A comprehensive review of syndromic forms of obesity: genetic etiology, clinical features, and molecular diagnosis. Current Obesity Reports, 13(2), 313-337.
- 7Chau, Y. Y., Liang, H., Tung, W. L., Hor, C. H. H., & Aik, W. S. (2025). Structural basis for Rab23 activation and a loss-of-function mutation in Carpenter syndrome. Journal of Biological Chemistry, 301(1).
- 8Utria, A. F., Mundinger, G. S., Bellamy, J. L., Zhou, J., Ghasemzadeh, A., Yang, R., … & Dorafshar, A. H. (2015). The importance of timing in optimizing cranial vault remodeling in syndromic craniosynostosis. Plastic and reconstructive surgery, 135(4), 1077-1084.
