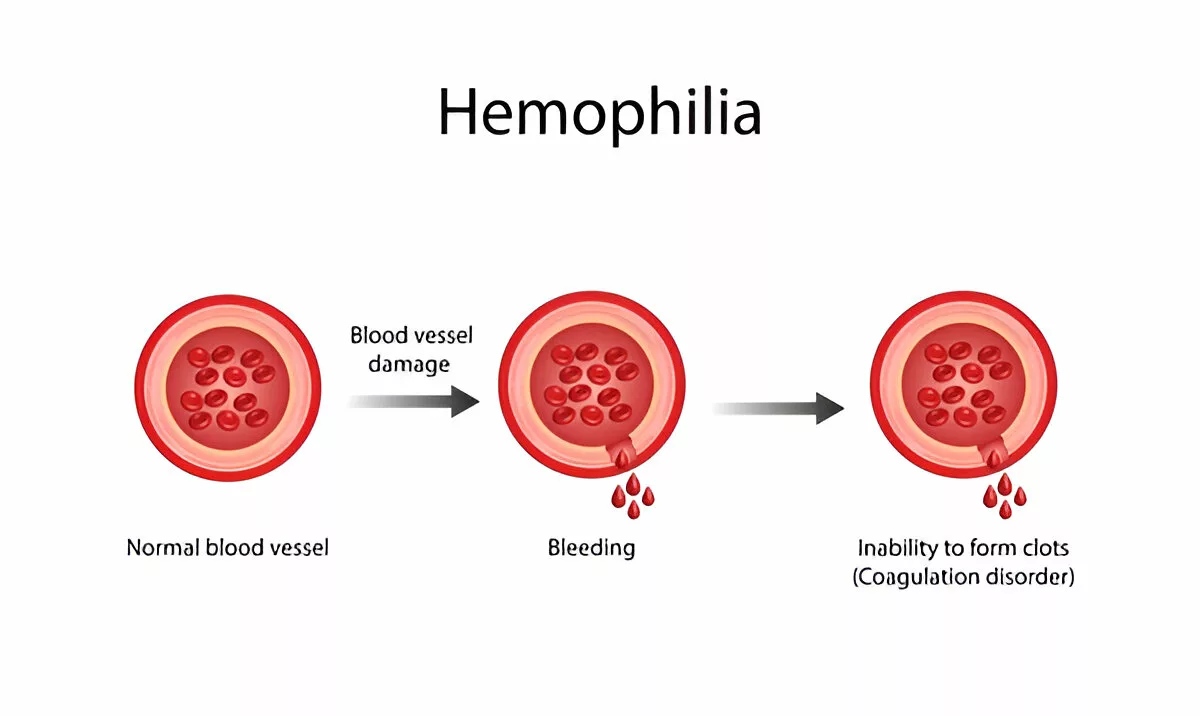
Hemophilia is a rare, inherited bleeding disorder where the blood doesn’t clot properly due to the absence or low levels of specific clotting factors. In individuals without hemophilia, when bleeding occurs, the body quickly forms clots using proteins called clotting factors to stop the blood loss. However, people with hemophilia lack one of these crucial factors, which means their blood can’t clot effectively. As a result, even minor injuries may cause prolonged bleeding, leading to serious health complications such as internal bleeding that can damage organs, joints, and surrounding tissues.1Srivastava A, Santagostino E, Dougall A, et al. (2020). WFH Guidelines for the Management of Hemophilia. Haemophilia, 26(Suppl 6):1–158. https://doi.org/10.1111/hae.14046
Hemophilia mainly affects males since it is typically inherited in an X-linked recessive pattern. Females can be carriers of the defective gene and usually don’t show symptoms, but in rare cases, they may experience bleeding issues if both of their X chromosomes carry mutations or if one X chromosome becomes inactivated more than the other in a process known as skewed X-inactivation. This can cause the faulty gene to become more dominant.2Bolton-Maggs PHB, Pasi KJ. (2003). Haemophilias A and B. Lancet, 361(9371):1801–1809. https://doi.org/10.1016/S0140-6736(03)13405-8
Historical Perspective:
Hemophilia has existed as a known medical condition for centuries. It was called “the royal disease” because of its presence in European royal descendants of Queen Victoria. In earlier times, limited medical knowledge meant that even minor injuries could prove fatal. However, thanks to major advancements in diagnostics and treatment, many individuals with hemophilia today live longer, healthier lives with better quality of care.3Tuddenham EGD. (1993). The molecular genetics of hemophilia A and B. Archives of Pathology & Laboratory Medicine, 117(1):80–85.
Understanding Blood Clotting
How does blood clotting normally work? When you get a cut, your body passes through a complex process involving platelets and clotting factors. Platelets rush to the site of injury and form a temporary plug. Meanwhile, clotting factors, specialized proteins in your blood, work together in a chain reaction to form a clot.
This entire system is called the coagulation cascade. It involves over a dozen clotting factors (named I to XIII), each activating the next in line. If any of these factors are missing or defective, as in the case of hemophilia, the chain breaks, and the clot doesn’t form properly.4Hoffman M, Monroe DM. (2001). A cell-based model of hemostasis. Thrombosis and Haemostasis, 85(6):958–965.
For example, in Hemophilia A, there’s a deficiency in factor VIII. In Hemophilia B, it’s factor IX that’s missing. Without these essential proteins, the clotting process stalls, and bleeding continues.
Types of Hemophilia
Hemophilia is not a one-size-fits-all condition. There are different types, mainly categorized based on which clotting factor is deficient. Knowing the type is crucial because it dictates the treatment and management approach.
Hemophilia A:
Hemophilia A is the most common type, accounting for about 80% of all cases. It’s caused by a deficiency in clotting factor VIII. People with severe Hemophilia A might experience spontaneous bleeding into joints and muscles without any injury. Those with milder forms might only notice symptoms during surgeries or major traumas.5Mannucci PM, Tuddenham EGD. (2001). The hemophilias—from royal genes to gene therapy. New England Journal of Medicine, 344(23):1773–1779. https://doi.org/10.1056/NEJM200106073442307
Hemophilia B:
Also known as Christmas disease (named after the first patient diagnosed), Hemophilia B results from a deficiency in factor IX. Factor IX molecules differ structurally from factor VIII, so specific concentrates must be used for each type.
Hemophilia C:
Much rarer than A or B, Hemophilia C involves a deficiency in factor XI. Unlike the others, Hemophilia C affects both males and females equally. It’s a milder form, and usually, spontaneous bleeding is uncommon in it. Most individuals with Hemophilia C only bleed excessively after surgery or trauma.6Salomon O, Steinberg DM, Seligsohn U. (2006). Variable bleeding manifestations characterize different types of human factor XI deficiency. Blood, 107(11):4621–4626. https://doi.org/10.1182/blood-2005-11-4557
Hemophilia A vs B
Here is a comparison of Hemophilia A and B that is essential for accurate diagnosis and treatment.
| Feature | Hemophilia A | Hemophilia B |
| Clotting Factor | Factor VIII deficiency | Factor IX deficiency |
| Prevalence | More common | Less common |
| Treatment | Factor VIII replacement therapy | Factor IX replacement therapy |
| Genetic Mutation | F8 gene | F9 gene |
Both types require similar management strategies but differ in the specific clotting factor involved.
Causes of Hemophilia
1. Genetic Mutations:
The most common cause of hemophilia is inherited genetic mutations in the genes that produce clotting factors VIII (Hemophilia A) or IX (Hemophilia B). These mutations disrupt the production or functionality of these proteins, resulting in impaired clotting ability.
2. Spontaneous Mutations:
Interestingly,Up to one-third of new hemophilia cases occur without a family history due to de novo mutations, which are spontaneous genetic changes that arise for the first time in an individual rather than being inherited from a parent.7Gitschier J, Wood WI, Goralka TM, et al. (1984). Characterization of the human factor VIII gene. Nature, 312:326–330.
3. Acquired Hemophilia:
Though extremely rare, acquired hemophilia occurs when the body’s immune system mistakenly attacks its clotting factors, particularly factor VIII. This form isn’t inherited and can occur in both men and women, often later in life or linked to autoimmune conditions, cancer, or certain medications.8Collins PW. (2011). Management of acquired hemophilia A. Journal of Thrombosis and Haemostasis, 9(Suppl 1):226–235. https://doi.org/10.1111/j.1538-7836.2011.04374.x
Hemophilia Symptoms
Some people might live most of their lives undiagnosed until a major surgery or injury reveals the disorder. However, those with severe forms will likely show symptoms early in life, even during infancy.
Common Symptoms:
- Prolonged Bleeding: This is often the first noticeable sign. After a cut, it may take longer than normal for the bleeding to stop. The same applies to internal bleeding, which is much harder to notice immediately.
- Spontaneous Bleeding: Individuals may experience bleeding without any obvious injury. This usually occurs in muscles or joints and is a hallmark of severe hemophilia.
- Excessive Bruising: Frequent and large bruises, even from minor bumps, can signal a bleeding disorder.
- Joint Pain and Swelling: Repeated bleeding into joints (hemarthrosis) can cause swelling, pain, and reduced mobility.9Gilbert MS. (2003). Prophylaxis: musculoskeletal evaluation. Haemophilia, 9(Suppl 1):9–12. https://doi.org/10.1046/j.1365-2516.9.s1.3.x
Severe vs. Mild Hemophilia Symptoms:
In severe hemophilia, symptoms can appear within the first year of life. Circumcision, vaccinations, or even teething produce excessive bleeding in newborns. The body suffers from dangerous internal bleeding episodes frequently in this condition.
Mild hemophilia, on the other hand, might go unnoticed until a surgery, dental procedure, or injury occurs. These individuals do not bleed spontaneously but will have prolonged bleeding when injured.
Symptoms in Children:
Parents may notice the following symptoms in their infants and toddlers:
- Excessive bruising from normal activities like crawling or bumping into things
- Unexplained swelling, particularly in joints
- Unusual bleeding after vaccinations or teething
- The children show irritability or refuse to move a limb (a sign of joint or muscle bleeding)
- Frequent nosebleeds
Encourage children to get regular exercise, yet parents need to watch for any physical harm and internal bleeding signs.
How is Hemophilia Diagnosed?
Proper diagnosis is the foundation for successful management. Medical tests become essential to detect signs, because early treatment can prevent serious complications like joint damage.
Blood Tests & Factor Assays:
The first step in diagnosis involves a series of blood tests to evaluate the blood clotting ability. The clotting factor assay is the most specific test that measures the activity levels of factors VIII and IX10Blanchette VS, Key NS, Ljung LR, et al. (2014). Definitions in hemophilia: communication from the SSC of the ISTH. Journal of Thrombosis and Haemostasis, 12(11):1935–1939. https://doi.org/10.1111/jth.12672
If the clotting time is prolonged, additional tests determine which factor is deficient. Doctors use these results to classify hemophilia as mild, moderate, or severe.
- Mild: 5%–40% of normal factor activity
- Moderate: 1%–5% of normal
- Severe: Less than 1%
Prenatal Testing:
In families who already know that they carry the hemophilia gene, prenatal diagnosis is possible. The two prenatal diagnostic tests, called chorionic villus sampling (CVS) or amniocentesis, can detect whether a fetus carries the gene mutation.11Goodeve AC. (2015). Hemophilia A and hemophilia B: molecular insights. Molecular Pathology, 68(5):245–252.
This information enables parents to prepare their baby’s care, while arranging specialist support at birth, and exploring all available choices.
Family History & Genetic Screening:
Because hemophilia is a genetic disorder, the diagnosis requires reviewing family medical history. If there are male relatives with unexplained bleeding problems or known hemophilia, it leads doctors to perform early newborn screening tests.
Medical professionals should offer genetic counseling to families who need to understand inheritance patterns and risks, particularly for carrier individuals.
Hemophilia Inheritance Pattern
Hemophilia typically follows an X-linked recessive inheritance pattern, which means the mutated gene responsible is located on the X chromosome. Since males have one X and one Y chromosome, inheriting a defective X from their mother results in the disease. Females, with two X chromosomes, are usually carriers.
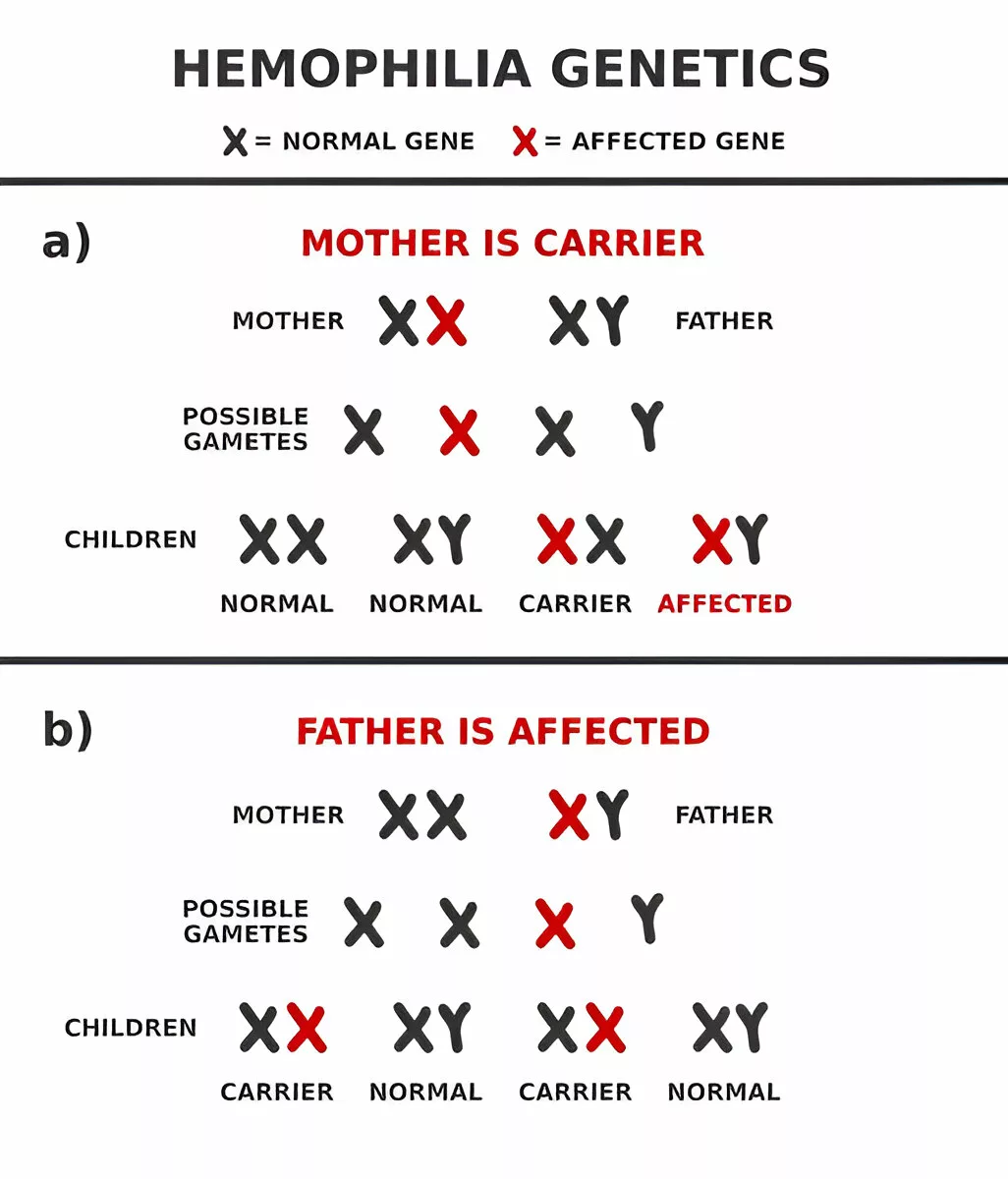
X-linked Recessive Inheritance:
Here’s how it works:
- A carrier mother has a 50% chance of passing the defective gene to her sons (who will have hemophilia) and a 50% chance of passing the gene to her daughters (who will become carriers themselves).
- A father with hemophilia can’t pass the disorder to his sons (he gives them a Y chromosome), but all his daughters will be carriers.
Here’s a simplified chart:
| Parent Status | Children Outcome |
| Carrier Mother + Healthy Father | 50% Carrier Daughters, 50% Affected Sons |
| Hemophiliac Father + Healthy Mother | 100% Carrier Daughters, 0% Affected Sons |
Carrier Mothers & Affected Sons:
Most often, mothers carry the defective gene without showing symptoms themselves. But they can pass it to their sons. A son born with hemophilia is often the first noticeable sign that the gene is present in a family, especially if there’s no known history.
Some carrier women do experience mild symptoms due to a phenomenon called skewed X-inactivation, where the healthy X chromosome is inactivated more often, reducing the amount of clotting factor they produce.
Rare Cases in Females:
Though hemophilia mostly affects males, females can be affected in rare cases, especially if:
- They inherit defective X chromosomes from both parents (a father with hemophilia and a carrier mother).
- They have Turner Syndrome (only one X chromosome, and it carries the mutation).
- They have a spontaneous mutation, and X-inactivation reduces the production of clotting factors.
Affected females may experience symptoms similar to mild hemophilia, such as heavy menstrual bleeding, nosebleeds, and bleeding after surgery.12Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, et al. (2006). Bleeding in carriers of hemophilia. Blood, 108(1):52–56. https://doi.org/10.1182/blood-2005-09-3726
Hemophilia Treatment Options
Thanks to modern medicine, hemophilia is no longer a fatal diagnosis. With appropriate and timely treatment, people with hemophilia can lead active and fulfilling lives.
Replacement Therapy:
The cornerstone of hemophilia treatment is replacement therapy, which involves infusing the missing clotting factor (either factor VIII or IX) into the bloodstream. This can be done:
- On-demand: Given only when a bleeding episode occurs.
- Prophylactically: Regular infusions to prevent bleeding, especially in severe cases.
Replacement therapy can use two types of products:
- Plasma-derived concentrates: Extracted from donated human blood13Franchini M, Mannucci PM. (2012). Plasma-derived vs recombinant factor VIII concentrates for the treatment of haemophilia A: recombinant is better. Blood Transfusion, 10(4):374–377.
- Recombinant factor products: Genetically engineered and free from human blood, reducing the risk of infections.
While effective, one challenge with replacement therapy is the development of inhibitors—antibodies that attack the infused factor and reduce its effectiveness. This complication requires specialized management with bypassing agents or immune tolerance therapy.
Non-Factor Therapies:
Newer treatment options are revolutionizing care, especially for those with inhibitors. One such breakthrough is emicizumab, a monoclonal antibody for Hemophilia A that mimics the function of factor VIII without being the actual protein. It’s administered via subcutaneous injection and can be given weekly or biweekly, making life much easier for patients.14Oldenburg J, Mahlangu JN, Kim B, et al. (2017). Emicizumab prophylaxis in hemophilia A with inhibitors. New England Journal of Medicine, 377:809–818. https://doi.org/10.1056/NEJMoa1703068
Other promising therapies include RNA interference drugs that increase the body’s natural ability to clot or bispecific antibodies that bridge components of the clotting cascade.
Living Well with Hemophilia
Living with hemophilia is about more than managing bleeds—it’s a continuous journey that touches every aspect of life: physical, emotional, and social. But here’s the truth: with the right strategies, support, and mindset, individuals with hemophilia can thrive.
Daily Management:
Managing a chronic condition like hemophilia requires preparation and vigilance. Bleeds, especially internal ones, can strike without warning. That’s why individuals must stay equipped and informed. Key strategies include:
- Self-infusion training – Patients can be learned at home to administer clotting factor. This can empower independence and ensure timely treatment.
- Recognizing early symptoms – Tingling, warmth, or stiffness in joints often precede internal bleeding. Quick action prevents damage.
- Wearing medical IDs – Wearing a bracelet or wallet card alerts emergency responders to your condition in case of an accident.
- Consistency with prophylaxis – Skipping preventive treatments increases the risk of joint issues, infections, and prolonged recovery times.
Preparedness is not just about physical health—it builds confidence and peace of mind.
Adapting to Everyday Life:
People with hemophilia don’t have to live an isolated life. With some adjustments, they can enjoy school, work, exercise, and even travel:
- Exercise: Staying active keeps joints strong. Opt for low-impact sports like swimming, cycling, walking, or yoga. Contact sports? Not ideal—but with supervision, even these can sometimes be modified.
- Travel: Yes, travel is doable. Keep a doctor’s letter, necessary medicines in your bag. Some use small coolers for factor that requires refrigeration. And you should have the knowledge of the nearest hospital, where you are going.
- School & Work: Kids can attend regular schools—just inform teachers and staff about the condition. Adults should seek careers that avoid high physical risks, like tech, creative fields, or office-based roles. Transparency with employers fosters a safer, more accommodating work environment.
Mental Health & Emotional Resilience:
Let’s be real—living with hemophilia isn’t just about the body. It takes a toll on the heart and mind, too. The unpredictability, limitations, and constant vigilance can trigger anxiety, stress, and even depression. It’s okay to feel overwhelmed.15Khair K, Holland M, Pollard D. (2013). The experience of girls and young women with bleeding disorders. Haemophilia, 19(6):e276–e281. https://doi.org/10.1111/hae.12168You’re not alone.
- Common challenges: Isolation, low self-esteem, fear of injuries, and missing out on social or physical activities.
- Coping strategies: Mindfulness, journaling, setting realistic goals, staying physically active (safely), and celebrating small wins, like an infusion done solo or a week without a bleed.
- Counseling helps: Therapists who specialize in chronic illness can help process fears, manage stress, and strengthen family communication.
With the right tools, people with hemophilia can become some of the most adaptable and self-aware individuals out there.
Support Systems That Matter:
Across the globe, organizations, treatment centers, and communities exist solely to help.
- Hemophilia Treatment Centers (HTCs) – They offer coordinated care: hematologists, physiotherapists, psychologists, and social workers—all in one place.
- Organizations like:
- The World Federation of Hemophilia (WFH)
- The National Hemophilia Foundation (NHF)
- Local groups, online forums, and advocacy communities
These spaces offer not only medical advice but also peer support, education, and occasionally financial assistance. Talking to someone who truly gets it can be life-changing.
Advances in Hemophilia Research
The landscape of hemophilia care is changing rapidly, with research for longer-lasting relief, easier administration, and potentially even a cure.
Gene Therapy:
Gene therapy is perhaps the most exciting advancement. It involves introducing a functional copy of the faulty gene into the patient’s liver cells, allowing the body to produce its clotting factor. For many, a single dose of gene therapy could reduce or eliminate the need for regular infusions.
Long-acting Factor Replacements:
Researchers are also developing longer-lasting factor concentrates that require fewer infusions. These extended half-life products remain in the bloodstream longer, reducing the frequency of treatment from several times a week to just once a week, or even less often.
This innovation improves adherence, especially for children and busy adults who struggle with traditional therapy schedules.
Future Prospects:
Looking ahead, the goal is not just to manage hemophilia but to normalize the lives of those who have it. Ongoing trials focus on:
- Oral medications that replace clotting factors
- More potent gene-editing tools like CRISPR16Nguyen TH, Anegon I. Successful correction of hemophilia by CRISPR/Cas9 genome editing in vivo: delivery vector and immune responses are the key to success. EMBO Mol Med. 2016 May 2;8(5):439-41. doi: 10.15252/emmm.201606325. PMID: 27138565; PMCID: PMC5130315.
- Therapies that reduce the immune response to clotting factor infusions
With continued research and funding, the future holds immense promise for people with hemophilia.
Conclusion
Medical progress, together with improved treatment options and community awareness, has turned hemophilia into a manageable chronic disease. Still, it’s not without its challenges. Still, it’s not without its challenges. From understanding clotting mechanics to daily life and coping with emotional stress, hemophilia affects every aspect of an individual’s life.
newly diagnosed with the condition and their parents, as well as anyone interested in learning more. Knowledge of hemophilia is the first step toward better care, empathy, and empowerment for all groups, including patients who receive new diagnoses and their parents, and anyone interested in learning about this condition. The combination of education and support allows people with hemophilia to build fulfilling lives.
Remember, knowledge is your most powerful tool. You should maintain awareness of your situation while being ready to seek assistance whenever needed. Because with the right care, hemophilia doesn’t have to hold anyone back.
Refrences
- 1Srivastava A, Santagostino E, Dougall A, et al. (2020). WFH Guidelines for the Management of Hemophilia. Haemophilia, 26(Suppl 6):1–158. https://doi.org/10.1111/hae.14046
- 2Bolton-Maggs PHB, Pasi KJ. (2003). Haemophilias A and B. Lancet, 361(9371):1801–1809. https://doi.org/10.1016/S0140-6736(03)13405-8
- 3Tuddenham EGD. (1993). The molecular genetics of hemophilia A and B. Archives of Pathology & Laboratory Medicine, 117(1):80–85.
- 4Hoffman M, Monroe DM. (2001). A cell-based model of hemostasis. Thrombosis and Haemostasis, 85(6):958–965.
- 5Mannucci PM, Tuddenham EGD. (2001). The hemophilias—from royal genes to gene therapy. New England Journal of Medicine, 344(23):1773–1779. https://doi.org/10.1056/NEJM200106073442307
- 6Salomon O, Steinberg DM, Seligsohn U. (2006). Variable bleeding manifestations characterize different types of human factor XI deficiency. Blood, 107(11):4621–4626. https://doi.org/10.1182/blood-2005-11-4557
- 7Gitschier J, Wood WI, Goralka TM, et al. (1984). Characterization of the human factor VIII gene. Nature, 312:326–330.
- 8Collins PW. (2011). Management of acquired hemophilia A. Journal of Thrombosis and Haemostasis, 9(Suppl 1):226–235. https://doi.org/10.1111/j.1538-7836.2011.04374.x
- 9Gilbert MS. (2003). Prophylaxis: musculoskeletal evaluation. Haemophilia, 9(Suppl 1):9–12. https://doi.org/10.1046/j.1365-2516.9.s1.3.x
- 10Blanchette VS, Key NS, Ljung LR, et al. (2014). Definitions in hemophilia: communication from the SSC of the ISTH. Journal of Thrombosis and Haemostasis, 12(11):1935–1939. https://doi.org/10.1111/jth.12672
- 11Goodeve AC. (2015). Hemophilia A and hemophilia B: molecular insights. Molecular Pathology, 68(5):245–252.
- 12Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, et al. (2006). Bleeding in carriers of hemophilia. Blood, 108(1):52–56. https://doi.org/10.1182/blood-2005-09-3726
- 13Franchini M, Mannucci PM. (2012). Plasma-derived vs recombinant factor VIII concentrates for the treatment of haemophilia A: recombinant is better. Blood Transfusion, 10(4):374–377.
- 14Oldenburg J, Mahlangu JN, Kim B, et al. (2017). Emicizumab prophylaxis in hemophilia A with inhibitors. New England Journal of Medicine, 377:809–818. https://doi.org/10.1056/NEJMoa1703068
- 15Khair K, Holland M, Pollard D. (2013). The experience of girls and young women with bleeding disorders. Haemophilia, 19(6):e276–e281. https://doi.org/10.1111/hae.12168
- 16Nguyen TH, Anegon I. Successful correction of hemophilia by CRISPR/Cas9 genome editing in vivo: delivery vector and immune responses are the key to success. EMBO Mol Med. 2016 May 2;8(5):439-41. doi: 10.15252/emmm.201606325. PMID: 27138565; PMCID: PMC5130315.
