What is Pompe Disease?
Pompe disease, also referred to as Glycogen Storage Disease Type II (GSDII), is a rare genetic disorder characterized by muscle wasting and weakness, primarily affecting infants and adolescents. The disease develops due to a lack of the enzyme acid alpha-glucosidase (GAA). The deficiency of GAA causes an excessive buildup of sugar glycogen in the body, which leads to severe muscular disintegration. Pompe disease is classified within a category of conditions called lysosomal storage disorders (LSDs). Lysosomes are small compartments within cells and are crucial in recycling various substances. If left untreated, this condition can be fatal.
Earlier, it was believed that Pompe disease was a rare disorder with a global prevalence of 1 in 40,000 individuals. However, new reports reveal a higher global prevalence, i.e., 1 in every 23,232 individuals. Pompe disease prevalence in East African and Asian populations is considerably higher than in other regions.1Park, K. S. (2021). Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Molecular genetics and metabolism reports, 27, 100734.
What Causes Pompe Disease?
Pompe disease results from mutations in the GAA gene, which codes for the GAA enzyme. It is inherited in an autosomal recessive pattern, meaning both parents must pass on a mutated copy of the gene for the disease to manifest. Genes are the fundamental units of heredity and come in dominant and recessive forms. While a dominant gene can express itself even when paired with a recessive gene, both copies of the recessive gene are required for a recessive trait like acid maltase deficiency to occur in Pompe disease.
How Does It Develop?
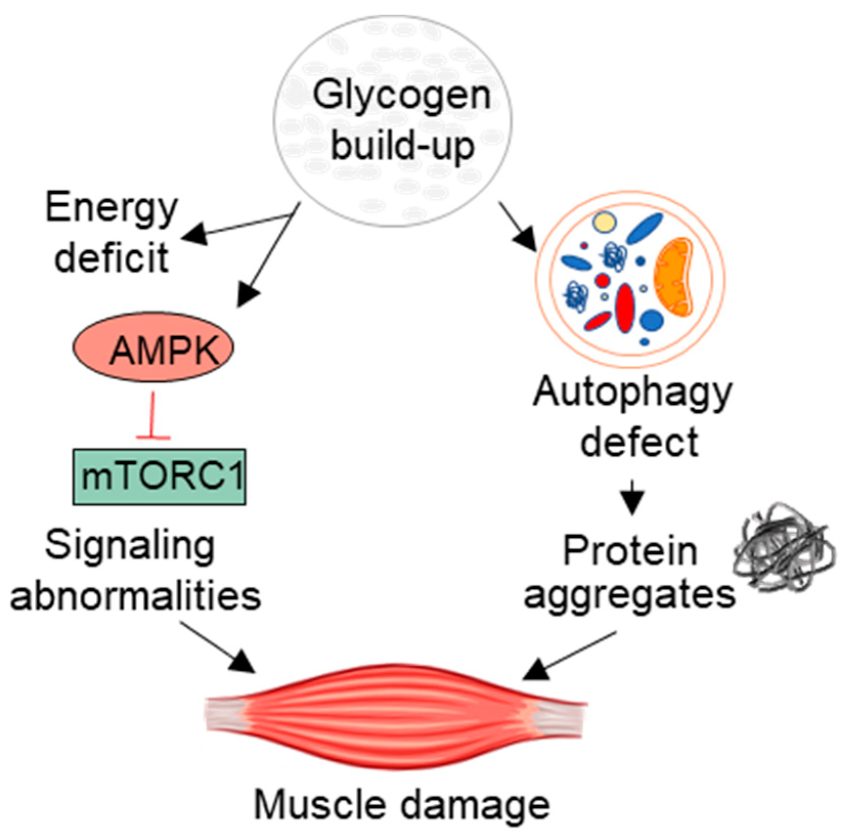
Clinicians classify Pompe disease as a glycogen storage disorder, a group of metabolic disorders characterized by abnormalities in glycogen synthesis and breakdown.
Glycogen is a complex carbohydrate (sugar) that the body stores in the liver and muscles.2Jensen, J., Rustad, P. I., & Lai, Y. C. (2011). The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Frontiers in physiology, 2, 13785. Glucose is another carbohydrate essential for providing energy throughout the body. The body gets glucose from the diet and utilizes it according to needs. However, the body converts the excess glucose into glycogen and stores it inside small compartments, i.e., lysosomes. These lysosomes are present within the muscles.
When the body’s energy requirement increases, specialized protein structures, such as enzymes, convert the stored glycogen back to glucose for energy.
Acid alpha-glucosidase (GAA) is an enzyme responsible for breaking down muscle glycogen. A deficiency of this enzyme causes a buildup of glycogen and consequent development of Pompe disease. Therefore, Pompe’s disease is also known as Acid maltase deficiency.
Lysosomes are intracellular compartments inside the muscles that recycle substances. In Pompe disease, glycogen accumulates within the lysosomes. Therefore, it is also known as lysosomal storage disease.
The absence or suboptimal levels of the GAA enzyme leads to glycogen accumulation in the muscles. This genetic infirmity mainly targets your skeletal muscles and the heart muscle. Unwanted glycogen breaks down the soft tissues and thus leads to serious complications.
Types Of Pompe Disease
Early/Infantile Onset Pompe Disease (IOPD)
This is the infantile form of Pompe disease, and it is generally more severe than the late-onset type. In this form of disease, very little to no enzyme production occurs. EOPD affects a wide array of muscles, leading to multiple complications.
Infantile disease causes significant enlargement of the heart, i.e., cardiomegaly. This symptom sets it apart from the latter type, as cardiomyopathy (heart muscle pathology) is usually not seen in the late-onset type.
Early onset Pompe disease symptoms are usually absent at birth and become evident at around four months. Such infants require special medical attention as symptoms progress quickly. Most patients cannot even celebrate their first birthdays without proper identification and treatment.
Life Expectancy: Classic Vs Non-Classic
As mentioned above, the main type of infantile form causes death within the first year of life. It is known as the classic infantile type. However, a milder form also exists, showing symptoms within the first six months, and survival is over two years. It is known as the non-classic infantile form.
Childhood/Juvenile Form
Sometimes, clinicians categorize Pompe disease into another type, i.e., the juvenile form. The symptoms are similar to the non-classic infantile type, and death ensues by the end of the third decade of life.3Di Rocco, M., Buzzi, D., & Tarò, M. (2007). Glycogen storage disease type II: clinical overview. Acta myologica, 26(1), 42.
Late Onset Pompe Disease (LOPD)
The adult-onset type can develop between one and ten years or into childhood. Compared to EOPD, the progression is slower and without cardiac involvement. Unlike the infant type, there is a partial deficiency of the GAA enzyme rather than a complete absence.
The patient gradually falls into the pit of death as muscle weakness engulfs the respiratory structures.
Pompe Disease Symptoms
Pompe disease symptoms depend on two factors:
- The severity of the disease: The extent of enzyme deficiency and myopathy determines the severity of the disease.
- Age of onset: Infants experience wider and more severe symptoms than children.
- Rate of progression: Disease’s progression rate varies from patient to patient.
There are some differences in symptoms of early-onset and late-onset Pompe disease. First, we shall discuss the symptoms common to both types and then move on to individual symptoms.
Frequent Respiratory Infections
Poor lung function and weak respiratory muscles hinder proper clearance of the airway passages. Lodging of microbes in the breathing pathway causes recurrent respiratory infections. This phenomenon is more pronounced in LOPD patients.
Hearing Difficulties
The glycogen storage disorder contributes to cochlear (inner ear) pathology. This consequently leads to hearing impairments.4van der Beek, N. A., Verschuure, H., Reuser, A. J., van der Ploeg, A. T., van Doorn, P. A., & Poublon, R. M. (2012). Hearing in adults with Pompe disease. Journal of Inherited Metabolic Disease, 35, 335-341.Sensorineural hearing loss is a significant sequelae of enzyme deficiency disorder. The intensity of impairment varies between individuals.5Hsueh, C. Y., Huang, C. Y., Yang, C. F., Chang, C. C., Lin, W. S., Cheng, H. L., … & Niu, D. M. (2021). Hearing characteristics of infantile-onset Pompe disease after early enzyme-replacement therapy. Orphanet Journal of Rare Diseases, 16, 1-8.
Swallowing/Feeding issues
Weakness of the oral and pharyngeal muscles interferes with swallowing. Infants have difficulty breastfeeding, while adolescents encounter dysphagia (inability to swallow).6Swift, G., Cleary, M., Grunewald, S., Lozano, S., Ryan, M., & Davison, J. (2017). Swallow prognosis and follow-up protocol in infantile-onset Pompe disease. JIMD Reports, Volume 33, 11-17.
Hepatomegaly
The liver is a major reserve for glycogen. Therefore, both Pompe disease types present with liver enlargement (hepatomegaly). The pathology of the body’s largest gland has serious consequences on generalized health. Hepatomegaly is more pronounced in the early-onset type.
Infantile-Onset Pompe Disease (IOPD)
Heart Enlargement
Cardiomegaly is an evident diagnostic feature of Pompe disease in infants. Thus, Dr. Pompe named the condition “cardiomegalia glycogenica” which translates to cardiac enlargement due to glycogen accumulation.7Limongelli, G., & Fratta, F. (2011). S1. 4 Cardiovascular involvement in Pompe disease. Acta Myologica, 30(3), 202. This leads to serious cardiac complications during early life.
Muscle Weakness And Loss Of Tone (Hypotonia)
Pompe disease has widespread muscle weakness. Myopathies are present in different muscles. Infants generally have frail bodies, and gaining weight seems impossible (due to their inability to thrive). In addition to weakness, it also decreases the tone of the muscles, leading to hypotonia. Pompe disease often makes infants appear like “rag dolls,” a muscle hypotonia condition called floppy infant syndrome. 8Howell, R. R., Byrne, B., Darras, B. T., Kishnani, P., Nicolino, M., & van der Ploeg, A. (2006). Diagnostic challenges for Pompe disease: an under-recognized cause of floppy baby syndrome. Genetics in Medicine, 8(5), 289-296.
Macroglossia
Tongue enlargement is a serious complication that lays the foundation for further issues (dysphagia, breathing issues, etc.). Glycogen’s infiltration into the tongue’s muscle fibers causes an increase in size.9Fatehi, F., Ashrafi, M. R., Babaee, M., Ansari, B., Sarraf, P., & Nafissi, S. (2021). Recommendations for infantile-onset and late-onset Pompe disease: an Iranian consensus. Frontiers in Neurology, 12, 739931.
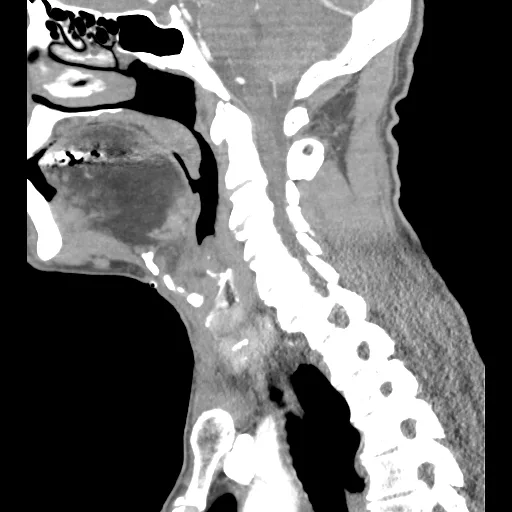
Case courtesy of Mackenna L. Senti, Radiopaedia.org. From the case rID: 147892
Breathing Insufficiency
Recurrent respiratory infections, cardiac pathologies, and tongue enlargement lead to breathing difficulties. Decreased lung capacity paired with heart pathologies causes early death.
Taking care of affected infants is a tough job. Frail children with enlarged hearts, livers, tongues, and weak breathing apparatus are difficult to manage for the parents.
Late-onset Pompe Disease (LOPD)
Children or adults experience symptoms later in life. Following is a list of the common LOPD symptoms:
Walking And Exercising Difficulties
In the initial stage, patients report an inability to exercise due to muscle weakness, especially in the arms and legs (limb-girdle weakness).10Lukacs, Z., Nieves Cobos, P., Wenninger, S., Willis, T. A., Guglieri, M., Roberts, M., … & Schoser, B. (2016). Prevalence of Pompe disease in 3,076 patients with hyperkalemia and limb-girdle muscular weakness. Neurology, 87(3), 295-298. Soon, the patient cannot walk, climb stairs, and even raise their arms. This leads to frequent falls and, consequently, a stagnant life.
Muscle Pain And Joint Stiffness
Frequently, skeletomuscular aches and stiffness accompany muscle weakness. Muscular pain over a large area and daytime tiredness keep patients from doing daily chores.
Dyspnea
In addition to muscle weakness and aches, patients encounter shortness of breath (dyspnea) with even the slightest exertion. Frequent lung infections and breathing difficulties prevent patients from engaging in all types of exercise.
Respiratory failure
As time progresses, children and adults start encountering problems in respiration. Muscle weakness involves the respiratory muscles, which gradually fail due to the impact of accumulated glycogen. Untreated respiratory failure eventually leads to death.11Shah, N. M., Sharma, L., Ganeshamoorthy, S., & Kaltsakas, G. (2020). Respiratory failure and sleep-disordered breathing in late-onset Pompe disease: a narrative review. Journal of Thoracic Disease, 12(Suppl 2), S235.
Spine Issues
Abnormal curvature of the spine is also a feature of Pompe disease. Doctors often notice the presence of spinal conditions like lumbar lordosis and scoliosis in patients.
Some patients also report morning headaches and irregular heart rhythms (arrhythmias) as consequences of Pompe’s disease.
Diagnosis
The first step in diagnosis involves taking the parents’ complete biological family history. The doctor correlates the medical history with the patient’s symptoms. As it is a genetic disease, family history plays an important role.
It might take a doctor weeks or even months to reach a diagnosis. This delay is partly due to overlapping symptoms of the disease with other muscular dystrophy conditions.
Screening Tests
A chest X-ray reveals evident cardiac enlargement (cardiomegaly) in infants. This is paired with an Electrocardiogram (ECG) to analyze the cardiac myopathy.
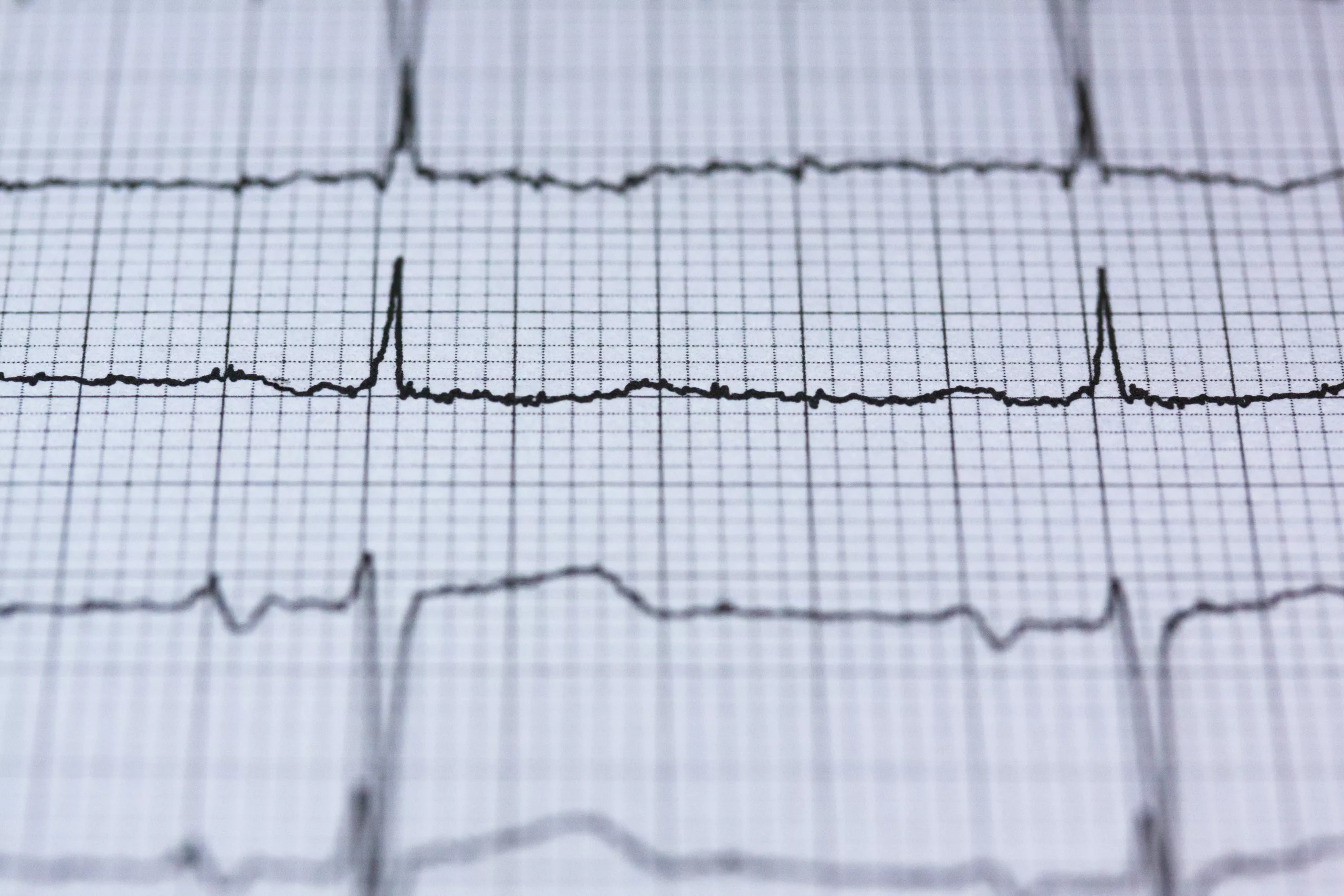
An ECG/EKG tests the electrical activity of the heart, and changes indicate irregularities. In the case of type Ⅱ glycogen storage disease, ECG shows short PR intervals and tall QRS complexes. Chest X-rays and ECGs are useful only in infantile forms.
Electromyography (EMG) is another diagnostic test that evaluates muscle activity. It tests the muscle’s response to electrical stimulation. A slower response indicates muscle weakness.
A lung (Pulmonary) Function test helps identify compromise in respiratory function. This test is especially important in diagnosing LOPD, where pulmonary failure is the main concern.
Your doctor may also advise a sleep study to evaluate changes during sleep.
Blood Tests
The Enzyme Assay measures enzyme (GAA) activity in the blood (or muscle via biopsy). IOPD patients have less than 1% normal enzyme levels, while LOPD patients have less than 40% normal enzyme levels.
Serum Creatine Kinase tests the levels of the enzyme creatine kinase. Elevated levels of CK in the blood are indicative of muscle damage. 2000 UI/L is the highest value noted for infantile-onset patients.
Damaged muscles may also raise serum levels of enzymes such as Aspartate aminotransferase (AST), Alanine aminotransferase (ALT), and Lactate dehydrogenase (LDH). Your doctor may also advise on the blood levels of the above-mentioned enzymes.
Pompe Disease Treatment
The most effective and FDA-approved treatment modality to manage this fatal disease is enzyme replacement therapy (ERT). A systematic review reveals that ERT positively impacts symptom alleviation, quality of life, and survival rate.12Dornelles, A. D., Junges, A. P. P., Pereira, T. V., Krug, B. C., Goncalves, C. B. T., Llerena Jr, J. C., … & Schwartz, I. V. D. (2021). A systematic review and meta-analysis of enzyme replacement therapy in late-onset Pompe disease. Journal of Clinical Medicine, 10(21), 4828.
Enzyme Replacement Therapy (ERT)
After an accurate diagnosis, the doctor will start intravenous (IV) enzyme replacement therapy. The most commonly used recombinant forms of the enzyme GAA for the treatment include:
- Alglucosidase alfa (Myozyme, Lumizyme)
- Avalglucosidase alfa-ngpt (Nexviazyme)
These genetically engineered medicines mimic the GAA enzyme in the body and convert the stored glycogen into usable forms. Myozyme treats infantile-onset disease (in babies and children), while Lumizyme can be prescribed to all ages. Nexviazyme is generally given to older adults suffering from late-onset Pompe disease.
A 2023 study revealed that high doses of alglucosidase alfa alleviated respiratory symptoms while increasing ventilator-free survival of infantile Pompe disease patients. 13Kishnani, P. S., Kronn, D., Suwazono, S., Broomfield, A., Llerena, J., Al-Hassnan, Z. N., … & Chien, Y. H. (2023). Higher dose alglucosidase alfa is associated with improved overall survival in infantile-onset Pompe disease (IOPD): data from the Pompe Registry. Orphanet Journal of Rare Diseases, 18(1), 381.
Patients receiving Alglucosidase alfa have almost five-fold lower mortality rates than untreated patients. Moreover, enzyme therapy improves survival, muscle function, and ambulation dependence.14Schoser, B., Stewart, A., Kanters, S., Hamed, A., Jansen, J., Chan, K., … & Toscano, A. (2017). Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. Journal of Neurology, 264, 621-630.
Complications Of Pompe Disease
Untreated infantile-onset disease ultimately leads to death owing to chronic breathing and heart problems. Adults with LOPD suffer from long-term muscle weakness. Therefore, many patients need walking assistance and oxygen support for breathing.
How To Live With It?
The disease requires you to keep an eye on the glycogen levels. Regular assessment is a must. As the symptoms of the disease are widespread, you need a multi-disciplinary approach for symptomatic management. Patients generally consult the following professionals to improve their quality of life:
- Cardiologists
- Speech therapists
- Nutrition therapists
- Physical therapists
Final Word
Pompe disease is a rare genetic disease caused by a mutation in the gene coding for the acid alpha-glucosidase enzyme (GAA). A lack of this enzyme leads to the accumulation of excess glycogen in the intracellular compartments (lysosomes) of muscles, which induces muscle damage. The infantile type affects infants and causes death within a year, while late-onset is present in children and adults. Patients suffer from skeletal muscle weakness, hepatomegaly cardiac enlargement, respiratory failure, and feeding/swallowing issues. Enzyme replacement therapy with alglucosidase alfa and avalglucosidase alfa-ngpt alleviates symptoms and increases survival. Symptomatic management involves treatment from nutrition, speech, and physical therapists.
Refrences
- 1Park, K. S. (2021). Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Molecular genetics and metabolism reports, 27, 100734.
- 2Jensen, J., Rustad, P. I., & Lai, Y. C. (2011). The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Frontiers in physiology, 2, 13785.
- 3Di Rocco, M., Buzzi, D., & Tarò, M. (2007). Glycogen storage disease type II: clinical overview. Acta myologica, 26(1), 42.
- 4van der Beek, N. A., Verschuure, H., Reuser, A. J., van der Ploeg, A. T., van Doorn, P. A., & Poublon, R. M. (2012). Hearing in adults with Pompe disease. Journal of Inherited Metabolic Disease, 35, 335-341.
- 5Hsueh, C. Y., Huang, C. Y., Yang, C. F., Chang, C. C., Lin, W. S., Cheng, H. L., … & Niu, D. M. (2021). Hearing characteristics of infantile-onset Pompe disease after early enzyme-replacement therapy. Orphanet Journal of Rare Diseases, 16, 1-8.
- 6Swift, G., Cleary, M., Grunewald, S., Lozano, S., Ryan, M., & Davison, J. (2017). Swallow prognosis and follow-up protocol in infantile-onset Pompe disease. JIMD Reports, Volume 33, 11-17.
- 7Limongelli, G., & Fratta, F. (2011). S1. 4 Cardiovascular involvement in Pompe disease. Acta Myologica, 30(3), 202.
- 8Howell, R. R., Byrne, B., Darras, B. T., Kishnani, P., Nicolino, M., & van der Ploeg, A. (2006). Diagnostic challenges for Pompe disease: an under-recognized cause of floppy baby syndrome. Genetics in Medicine, 8(5), 289-296.
- 9Fatehi, F., Ashrafi, M. R., Babaee, M., Ansari, B., Sarraf, P., & Nafissi, S. (2021). Recommendations for infantile-onset and late-onset Pompe disease: an Iranian consensus. Frontiers in Neurology, 12, 739931.
- 10Lukacs, Z., Nieves Cobos, P., Wenninger, S., Willis, T. A., Guglieri, M., Roberts, M., … & Schoser, B. (2016). Prevalence of Pompe disease in 3,076 patients with hyperkalemia and limb-girdle muscular weakness. Neurology, 87(3), 295-298.
- 11Shah, N. M., Sharma, L., Ganeshamoorthy, S., & Kaltsakas, G. (2020). Respiratory failure and sleep-disordered breathing in late-onset Pompe disease: a narrative review. Journal of Thoracic Disease, 12(Suppl 2), S235.
- 12Dornelles, A. D., Junges, A. P. P., Pereira, T. V., Krug, B. C., Goncalves, C. B. T., Llerena Jr, J. C., … & Schwartz, I. V. D. (2021). A systematic review and meta-analysis of enzyme replacement therapy in late-onset Pompe disease. Journal of Clinical Medicine, 10(21), 4828.
- 13Kishnani, P. S., Kronn, D., Suwazono, S., Broomfield, A., Llerena, J., Al-Hassnan, Z. N., … & Chien, Y. H. (2023). Higher dose alglucosidase alfa is associated with improved overall survival in infantile-onset Pompe disease (IOPD): data from the Pompe Registry. Orphanet Journal of Rare Diseases, 18(1), 381.
- 14Schoser, B., Stewart, A., Kanters, S., Hamed, A., Jansen, J., Chan, K., … & Toscano, A. (2017). Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. Journal of Neurology, 264, 621-630.
