Alagille syndrome is a genetic condition that primarily affects the liver but can also impact other organs, such as the heart, kidneys, and eyes. In this disease, bile builds up in the liver because there are too few bile ducts to drain it, a condition known as bile duct paucity. This can lead to liver damage and other complications. Alagille syndrome is a rare disease, affecting approximately one in every 70,000 newborns.1Johns Hopkins Medicine. (n.d.). Alagille syndrome. Retrieved July 7, 2024, from https://www.hopkinsmedicine.org/health/conditions-and-diseases/alagille-syndrome
What is Alagille Syndrome?
Alagille syndrome (ALGS) is a multisystemic genetic disorder primarily characterized by cholestatic liver disease, although it also affects various other organ systems. The clinical manifestations can vary significantly, even among members of the same family. It is inherited in an autosomal dominant pattern, meaning if one parent carries the mutation, there’s a 50% chance it will be passed to their children.
Most cases (approximately 70-90%) are caused by mutations in the JAG1 gene, while mutations in the NOTCH2 gene account for less than 2%. Nearly half of all cases arise from new mutations not inherited from parents.2Han Y, Zhu K, Wu H, Chen B, Hu S, Lai D, Tou J. Case Report: Novel JAG1 gene mutations in two infants with alagille syndrome characterized by cholestasis. Front Pediatr. 2022 Oct 20;10:1017647. doi: 10.3389/fped.2022.1017647. PMID: 36340723; PMCID: PMC9631024. It is alternatively referred to as arteriohepatic dysplasia, Alagille-Watson syndrome, Watson-Miller syndrome, or syndromic bile duct paucity.
This syndrome is defined by a lack of intrahepatic bile ducts, leading to bile buildup in the liver (cholestasis), combined with the presence of at least three of the following five clinical symptoms:
- Cholestasis
- Cardiac disorders
- Skeletal abnormalities
- Ocular disturbances
- Distinctive facial features
Signs & Symptoms of Alagile Syndrome
Many people with this syndrome have mild symptoms and can lead normal lives. Others may experience various symptoms, as Alagille syndrome is not restricted to the liver or heart alone. Many other body parts, like the thyroid, kidney, and brain, can also be affected. The symptoms include:
Liver-Related Symptoms
Individuals with Alagille syndrome often experience several liver-related symptoms due to impaired bile flow and liver function. Pruritus, or severe itchiness of the skin, occurs due to bile stasis. Jaundice is another common symptom, characterized by a yellowish discoloration of the skin and the whites of the eyes due to the accumulation of bilirubin in the blood. Additionally, patients may notice a darkening of urine and lightening of stools, which are caused by reduced bile flow affecting the normal excretion of bilirubin and bile pigments. Xanthomas, yellowish deposits of fat that appear as papules, nodules, or plaques on the skin, can also occur. These are often associated with lipid disorders and result from the high levels of cholesterol and triglycerides seen in liver disease.
Cardiac Defects:
Many heart problems involve heart murmurs and heart defects. The most common defect in the heart is peripheral pulmonary stenosis. Other main structural cardiac defects include tetralogy of Fallot and patent ductus arteriosus.
Dysmorphic Features:
A prominent forehead, deep-set or sunken eyes, a straight nose with a flattened tip, and a pointed chin.
Skeletal Abnormalities:
Vascular Involvement:
Alagille syndrome can involve vascular anomalies, including narrowing (stenosis) of the pulmonary arteries and other vessels. While less common, there can be abnormalities in the cerebral vessels, but swelling of vessel walls is not typical.
Kidney Involvement:
Individuals with Alagille syndrome may experience kidney abnormalities, including structural malformations and, occasionally, renal tubular acidosis. Vesicoureteral reflux may also occur but is not a primary feature of the syndrome.
Eye Abnormalities:
Posterior embryotoxon, a condition affecting the eye’s cornea, is observed in almost 95% of the cases, though it usually doesn’t impact vision. In this condition, there is an abnormal thickening and anterior displacement of the corneal ring (Schwalbe’s line) in the eye, often visible as a white line around the iris.3Porter, J. D. H., & Meenken, C. (1999). Posterior embryotoxon in Alagille syndrome. Ophthalmology, 106(4), 833-839. https://doi.org/10.1016/S0161-6420(99)90072-6
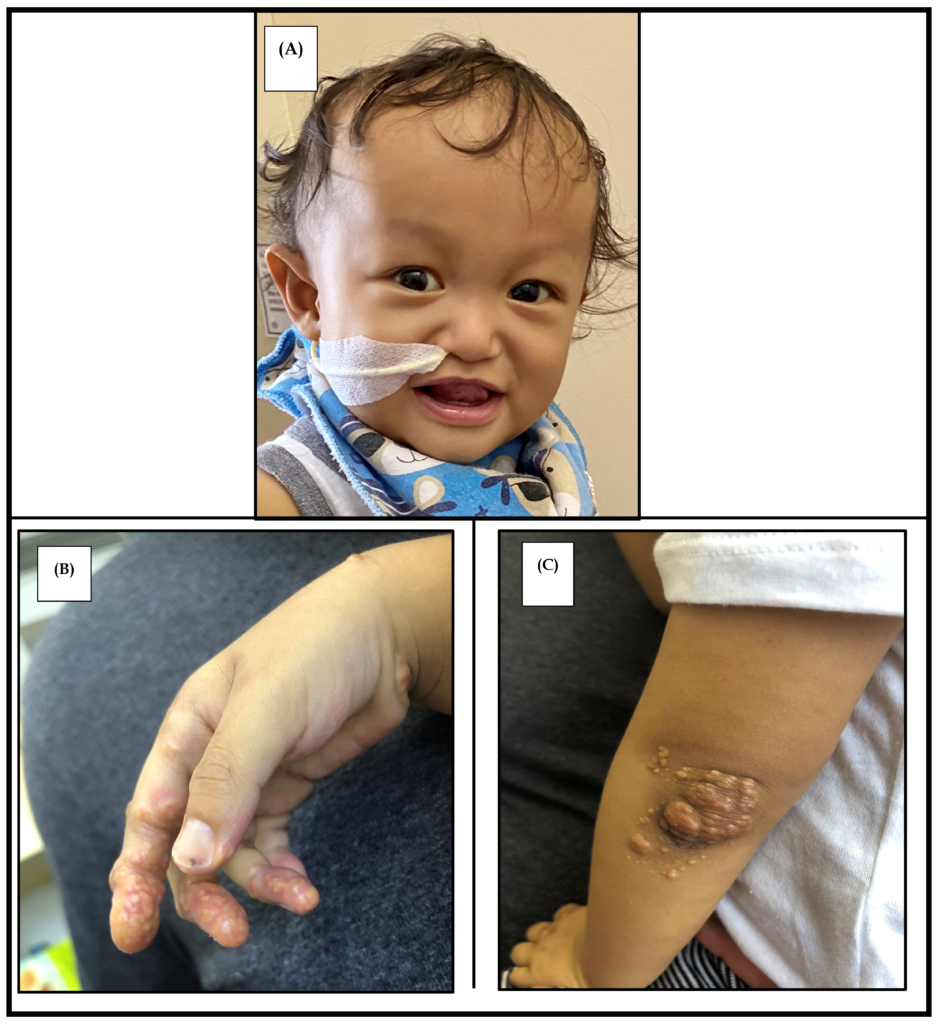
(A) Characteristic facies showing a prominent forehead, hypertelorism, straight nose with a bulbous tip, and a pointed chin. Photograph used with parental consent.
(B) Cutaneous xanthoma on the palmar surface of the hand.
(C) Xanthoma observed on the elbow. Image Courtesy: Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics. 2020; 10(11):907. https://doi.org/10.3390/diagnostics10110907 available under CC BY 4.0
What Causes Alagille Syndrome?
This syndrome is most importantly associated with genetic mutations. Mutations in JAG1 occur more than in NOTCH2. In more than 50% of the people with Alagille syndrome, the disease is caused by a new gene mutation, which means the gene mutation was not inherited from a parent. The mutations in JAG1 and NOTCH2 genes are the main cause of this syndrome.4Fischetto R, Palmieri VV, Tripaldi ME, Gaeta A, Michelucci A, Delvecchio M, Francavilla R, Giordano P. Alagille Syndrome: A Novel Mutation in JAG1 Gene. Front Pediatr. 2019 May 15;7:199. doi: 10.3389/fped.2019.00199. PMID: 31157196; PMCID: PMC6529843.
It is an autosomal dominant disease and is passed from parents to children. Children who have one parent with this syndrome have a 50 percent chance of inheriting the gene mutation and having the disease.
Diagnosis of Alagille Syndrome
Alagille syndrome is diagnosed based on clinical features and can be confirmed through various diagnostic procedures. Diagnosis typically requires the presence of at least three out of seven major clinical features, which may include:5Diaz-Frias J, Kondamudi NP. Alagille Syndrome. [Updated 2023 Aug 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507827/
- Dysmorphic features
- Vertebral anomalies (66%)—A characteristic “butterfly” vertebrae may be observed but are of no structural significance6 Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis, and management. European Journal of Human Genetics: EJHG. Mar 2012;20(3):251-7. doi:10.1038/ejhg.2011.181
- Changes in heart and blood vessel structures or heart murmurs.
- Cholestasis
- Abnormality in kidney function or structure
- Xanthomas
- Posterior Embryotoxon
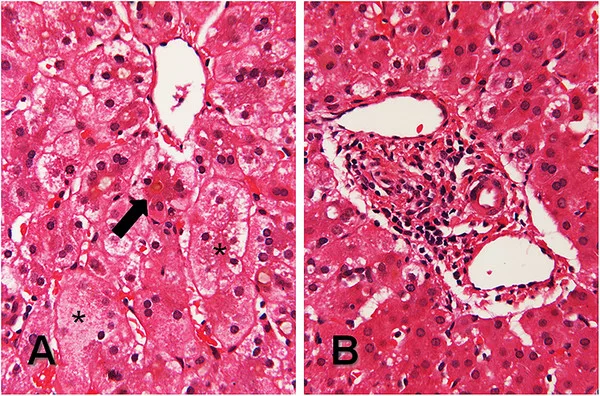
Also, genetic testing that can identify mutations in the JAG1 or NOTCH2 genes associated with Alagille syndrome helps confirm the diagnosis. Liver biopsy, once considered essential, is now less necessary due to advancements in diagnostic criteria. Cholestasis, on histology, serves as a key indicator instead of bile duct paucity.
Laboratory Testing:
The following laboratory abnormalities can be present:
- Hepatic dysfunction: LFTs will usually indicate elevated levels of:
o Direct bilirubin
o Serum aminotransferases,
o Bile acids
o Cholesterol
o Triglycerides
o Gamma-glutamyl transpeptidase - Fat-soluble vitamin deficiencies
- Prolongation of prothrombin time (PT) or activated partial thromboplastin time (aPTT)
Other assessments, such as urine analysis for renal tubular acidosis, stool examination for pancreatic function, cardiac echocardiography, and skeletal assessment via X-rays, are also crucial for a thorough diagnosis.
Life Expectancy in Alagille syndrome
Some people with this syndrome can lead normal lives. About 75% of patients diagnosed in childhood can live for at least 20 years.7Rare Disease Advisor. (n.d.). Alagille syndrome prognosis. Retrieved July 7, 2024, from https://www.rarediseaseadvisor.com/disease-info-pages/alagille-syndrome-prognosis/#:~:text=As%20treatment%20options%20increase%2C%20the,the%20age%20of%2020%20years. Alagille syndrome presents itself with various symptoms and some people may experience only the effects related to one organ while others may have effects in multiple organs. Most of the patients with this syndrome only have mild symptoms that usually go away.
Management of Alagille Syndrome
Treatment for this syndrome is tailored to address each individual’s unique symptoms. It involves a collaborative effort among specialists such as pediatricians, gastroenterologists, cardiologists, ophthalmologists, and other healthcare providers to ensure comprehensive and systematic care planning.
Treatment of Cholestatic Liver Disease:
In managing liver disease associated with Alagille syndrome, treatment focuses on providing support to alleviate severe itching and reduce the accumulation of cholesterol with medications like ursodeoxycholic acid, naltrexone, rifampin, colesevelam, and cholestyramine.
Chronic pruritus is often resistant to treatment with medications, and it is a problem that involves the patient’s overall life quality. Research shows the beneficial influence of either cholestyramine (12.5-15g/day) or rifampin (as a second-line treatment) on managing bile acid-induced itching found in Alagille syndrome patients.8Hofmann A. F. (2002). Rifampicin and treatment of cholestatic pruritus. Gut, 51(5), 756–757. https://doi.org/10.1136/gut.51.5.756 Other drugs include:
Maralixibat
The IBAT inhibitor maralixibat (Livmarli) was approved in September 2021 as a release for the pruritic cholestasis treatment in patients with Alagille syndrome who were at least one year old.9Kamath, B. M., Goldstein, A., Howard, R., Garner, W., Vig, P., Marden, J. R., Billmyer, E., Anderson, A., Kirson, N., Jacquemin, E., & Gonzales, E. (2023). Maralixibat Treatment Response in Alagille Syndrome is Associated with Improved Health-Related Quality of Life. The Journal of Pediatrics, 252, 68–75.e5. https://doi.org/10.1016/j.jpeds.2022.09.001
Odevixibat
Another FDA-approved iBAT inhibitor, odevixibat (Bylvay), was also disclosed in June 2023, intended for adult patients aged one year and older with Alagille syndrome suffering from itching.10Ganschow, R., & Maucksch, C. (2023). Odevixibat Treatment of Alagille Syndrome: A Case Report. JPGN reports, 4(2), e301. https://doi.org/10.1097/PG9.0000000000000301
Endocarditis Prophylaxis:
A cardiologist should attend to the cases of clinically manifest cardiac diseases. All patients, except those with peripheral pulmonic stenosis, require subacute bacterial endocarditis prophylaxis.
Surgical Treatment:
The operative treatment for Alagille syndrome includes the following:
Partial Biliary Diversion:
This procedure alleviates symptoms of Alagille syndrome by interrupting the enterohepatic circulation of bile acids. It aims to reduce bile salt retention and associated symptoms such as pruritus (itchiness) by diverting bile from the liver to the intestine, which can help manage symptoms in some patients.11Sheflin-Findling S, Arnon R, Lee S, Chu J, Henderling F, Kerkar N, Iyer K. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012 Jul;47(7):1453-6. doi: 10.1016/j.jpedsurg.2012.04.008. PMID: 22813814.
Liver Transplantation:
Orthotopic liver transplantation is considered for individuals with Alagille syndrome who have refractory disease, meaning that their condition does not respond to other treatments.
Indications for consideration of liver transplantation include the following:
- Progressive hepatic dysfunction
- Severe portal hypertension
- Failure to thrive
- Intractable pruritus and osteodystrophy.
Compared to those who do not require liver transplantation, the 5-year survival rate for the ones who require hepatic transplantation is 60%.12Ghelichi-Ghojogh, M., Rajabi, A., Mohammadizadeh, F., Shojaie, L., Vali, M., Afrashteh, S., Hassanipour, S., Nikbakht, H. A., Khezri, R., Allah Kalteh, E., & Fararouei, M. (2022). Survival Rate of Liver Transplantation in Asia: A Systematic Review and Meta-Analysis. Iranian journal of public health, 51(10), 2207–2220. https://doi.org/10.18502/ijph.v51i10.10979 On the other hand, the survival rate is 80% for the groups that do not need their liver replaced by transplantation.
Cardiac Surgery
The most serious heart issues such as Tetralogy of Fallot, transposition of great vessels, Ventricular septal defect (VSD) with pulmonary atresia (PA), and failing to close the atrial septal defect (ASD) and ventricular septal defect (VSD) may require the patients to undergo the cardiac surgery at a later stage.
Genetic Counseling:
Since Alagille syndrome is genetic, genetic counseling is essential for families to understand the inheritance pattern and risks associated with future pregnancies.
Dietary Modifications:
In patients with Alagille syndrome, the diet should focus on properly absorbing carbohydrates and medium-chain triglycerides (MCTs) rather than long-chain fatty acids. MCTs are more easily absorbed by individuals with liver and bile duct issues, as they do not require bile for digestion. Breastfed babies can be supplemented with MCT oil to enhance caloric intake and improve nutrition.13National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). (n.d.). Eating, diet, & nutrition for Alagille syndrome. Retrieved July 7, 2024, from https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome/eating-diet-nutrition
Supplemental treatment with vitamins and nutrients is essential for individuals with malabsorption. A good vitamin dosage is important for optimal growth and development in patients with Alagille syndrome. Water-soluble forms of vitamins A, D, E, and K are poorly absorbed. Complexes of vitamins A, D, E, and K with polyethylene glycol compounds are generally well tolerated by patients and are better absorbed.
For those with significant malabsorption, cholestasis, or related complications, feeding options may include enteral nutrition through a nasogastric tube or gastrostomy. This can provide necessary nutrition when oral intake is insufficient due to gastrointestinal issues. In cases of severe malnutrition or failure to thrive, intravenous nutrition may be used temporarily to provide essential nutrients.
Alagille Syndrome Vs. Biliary Atresia
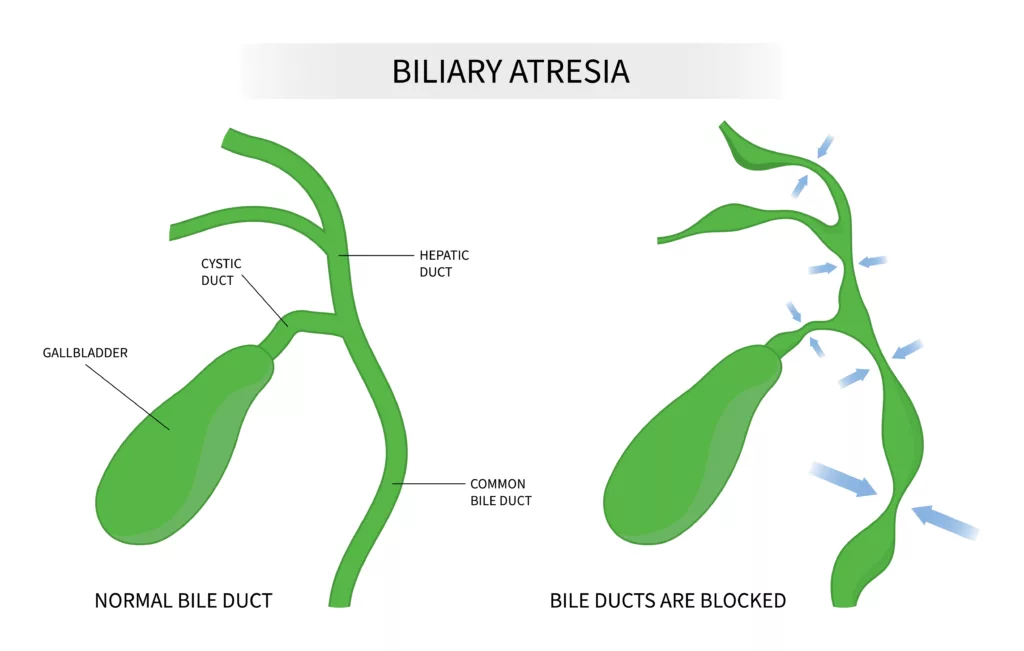
The pathogenesis of biliary atresia remains unclear, but genetic predisposition may play a role in some cases. Biliary atresia is a congenital condition in which the bile ducts that transport bile from the liver to the gallbladder are damaged or blocked. This obstruction leads to bile accumulation in the liver, causing damage and potentially progressing to liver cirrhosis and, eventually, liver failure. While the exact cause of biliary atresia is still under debate, it is thought to involve developmental defects and autoimmune processes.
Alagille syndrome, conversely, is a genetic disorder primarily affecting the liver and other organs due to mutations in the JAG1 or NOTCH2 genes. It may resemble biliary atresia, particularly in newborns and young children, making them sometimes difficult to distinguish. However, a key distinguishing feature is the presence of at least one additional symptom beyond cholestasis in Alagille syndrome, such as characteristic facial features, cardiac anomalies, or skeletal abnormalities.14Spinner NB, Loomes KM, Krantz ID, et al. Alagille Syndrome. 2000 May 19 [Updated 2024 Jan 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1273/
Prevention of Alagille Syndrome
Alagille syndrome is a genetic disorder involving multiple organs, mainly the liver. If the symptoms are mild, life expectancy is good, and further damage can be prevented. Experts have not yet discovered any way to avoid it, but managing the symptoms properly and visiting the doctor regularly can help prevent serious complications. Follow instructions for lifestyle changes and diet to improve health and comfort.
Conclusion
Alagille syndrome is a very rare genetic disorder with basic symptoms like the lack of bile ducts, which results in bile collection. Symptoms are not limited to specific organs like ears, heart, eyes, or kidneys. Genetic testing and clinical manifestation or symptoms diagnose the disorder. Therapy is divided into medical care, surgery, and liver transplantation. Early diagnosis remains the key factor for adequate treatment and a good prognosis.
Refrences
- 1Johns Hopkins Medicine. (n.d.). Alagille syndrome. Retrieved July 7, 2024, from https://www.hopkinsmedicine.org/health/conditions-and-diseases/alagille-syndrome
- 2Han Y, Zhu K, Wu H, Chen B, Hu S, Lai D, Tou J. Case Report: Novel JAG1 gene mutations in two infants with alagille syndrome characterized by cholestasis. Front Pediatr. 2022 Oct 20;10:1017647. doi: 10.3389/fped.2022.1017647. PMID: 36340723; PMCID: PMC9631024.
- 3Porter, J. D. H., & Meenken, C. (1999). Posterior embryotoxon in Alagille syndrome. Ophthalmology, 106(4), 833-839. https://doi.org/10.1016/S0161-6420(99)90072-6
- 4Fischetto R, Palmieri VV, Tripaldi ME, Gaeta A, Michelucci A, Delvecchio M, Francavilla R, Giordano P. Alagille Syndrome: A Novel Mutation in JAG1 Gene. Front Pediatr. 2019 May 15;7:199. doi: 10.3389/fped.2019.00199. PMID: 31157196; PMCID: PMC6529843.
- 5Diaz-Frias J, Kondamudi NP. Alagille Syndrome. [Updated 2023 Aug 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507827/
- 6Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis, and management. European Journal of Human Genetics: EJHG. Mar 2012;20(3):251-7. doi:10.1038/ejhg.2011.181
- 7Rare Disease Advisor. (n.d.). Alagille syndrome prognosis. Retrieved July 7, 2024, from https://www.rarediseaseadvisor.com/disease-info-pages/alagille-syndrome-prognosis/#:~:text=As%20treatment%20options%20increase%2C%20the,the%20age%20of%2020%20years.
- 8Hofmann A. F. (2002). Rifampicin and treatment of cholestatic pruritus. Gut, 51(5), 756–757. https://doi.org/10.1136/gut.51.5.756
- 9Kamath, B. M., Goldstein, A., Howard, R., Garner, W., Vig, P., Marden, J. R., Billmyer, E., Anderson, A., Kirson, N., Jacquemin, E., & Gonzales, E. (2023). Maralixibat Treatment Response in Alagille Syndrome is Associated with Improved Health-Related Quality of Life. The Journal of Pediatrics, 252, 68–75.e5. https://doi.org/10.1016/j.jpeds.2022.09.001
- 10Ganschow, R., & Maucksch, C. (2023). Odevixibat Treatment of Alagille Syndrome: A Case Report. JPGN reports, 4(2), e301. https://doi.org/10.1097/PG9.0000000000000301
- 11Sheflin-Findling S, Arnon R, Lee S, Chu J, Henderling F, Kerkar N, Iyer K. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012 Jul;47(7):1453-6. doi: 10.1016/j.jpedsurg.2012.04.008. PMID: 22813814.
- 12Ghelichi-Ghojogh, M., Rajabi, A., Mohammadizadeh, F., Shojaie, L., Vali, M., Afrashteh, S., Hassanipour, S., Nikbakht, H. A., Khezri, R., Allah Kalteh, E., & Fararouei, M. (2022). Survival Rate of Liver Transplantation in Asia: A Systematic Review and Meta-Analysis. Iranian journal of public health, 51(10), 2207–2220. https://doi.org/10.18502/ijph.v51i10.10979
- 13National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). (n.d.). Eating, diet, & nutrition for Alagille syndrome. Retrieved July 7, 2024, from https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome/eating-diet-nutrition
- 14Spinner NB, Loomes KM, Krantz ID, et al. Alagille Syndrome. 2000 May 19 [Updated 2024 Jan 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1273/
