
Glanzmann thrombasthenia (GT) is a bleeding disorder caused by impaired platelet function. It’s characterized by lifelong episodes of bleeding, often presenting as a spontaneous bleed after a minor trauma in childhood. GT is a rare disease, affecting approximately 1 in 1,000,000 people globally. Since patients typically have a normal platelet count and morphology, healthcare providers frequently misdiagnose it. Therefore, it’s important to understand this disorder and have a high index of suspicion, not only for accurate diagnosis, but also to prevent life-threatening bleeding episodes and manage patients effectively.
What is Glanzmann Thrombasthenia?
Glanzmann thrombasthenia (GT) is a rare autosomal recessive disorder of platelet function. It is caused by quantitative or qualitative defects in the platelet membrane glycoprotein αIIbβ3 complex, leading to impaired platelet aggregation.
(Note that Integrin αIIbβ3, also known as GP IIb/IIIa, is one of the five glycoproteins expressed on the platelet surface. It plays a crucial role in platelet aggregation and clot retraction in the event of any injury.
Pathophysiology of Glanzmann Thrombasthenia:
Normally, when a blood vessel ruptures, platelets rush to the site of injury and become activated. These activated platelets express αIIbβ3 receptors, which bind to soluble fibrinogen. This step allows for bridge formation amongst the platelets, helping them stick together to form the primary platelet plug. Apart from this, the platelet contractile machinery causes the platelet-fibrin clot to tighten, resulting in clot retraction.
In Glanzmann thrombasthenia, fibrinogen can’t bind to the αIIbβ3 receptor due to reduced expression or function of the αIIbβ3 complex. As a result, platelets are unable to aggregate to form the platelet plug.
Since αIIbβ3-mediated linkage to the clot is absent in GT, platelets can’t transmit contractile forces to reshape the clot. This results in weak or absent clot retraction, causing loose and unstable thrombi.1Nurden A. (2021). Profiling the Genetic and Molecular Characteristics of Glanzmann Thrombasthenia: Can It Guide Current and Future Therapies?. Journal of blood medicine, 12, 581–599. https://doi.org/10.2147/JBM.S273053. Although you have a normal platelet count with normal activation pathways, the formation of a hemostatic plug is severely impaired.
Classification of Glanzmann Thrombasthenia
On the level of expression and functional activity of the αIIbβ3 receptors, Glanzmann Thrombasthenia is classified into.
- Type I: In type I, the expression of αIIbβ3 is ≤5% as compared to normal. It is the most common type of GT, accounting for about 78% of the total cases. Patients show virtually no platelet aggregation and clot retraction in this type.2Botero, J. P., Lee, K., Branchford, B. R., Bray, P. F., Freson, K., Lambert, M. P., Luo, M., Mohan, S., Ross, J. E., Bergmeier, W., & Di Paola, J. (2020). Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica, 105(4), 888–894. https://doi.org/10.3324/haematol.2018.214239
- Type II: It is a less severe type of GT, in which 5-15% of αIIbβ3 are expressed. Patients of type II Glanzmann thrombasthenia also have impaired platelet aggregation, but the clot retraction is partially normal.3Nurden A. (2021). Profiling the Genetic and Molecular Characteristics of Glanzmann Thrombasthenia: Can It Guide Current and Future Therapies?. Journal of blood medicine, 12, 581–599. https://doi.org/10.2147/JBM.S273053
- Variant/Type III: Though the variant type has normal or near normal αIIbβ3 receptor expression, the receptors still can’t bind to fibrinogen. It is the least common, accounting for only 8% of the total GT cases.4Botero, J. P., Lee, K., Branchford, B. R., Bray, P. F., Freson, K., Lambert, M. P., Luo, M., Mohan, S., Ross, J. E., Bergmeier, W., & Di Paola, J. (2020). Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica, 105(4), 888–894. https://doi.org/10.3324/haematol.2018.214239
How common is Glanzmann Thrombasthenia?
GT is a rare disease. Its worldwide prevalence is 1 in 1,000,000 individuals, with some regions of the world having slightly higher prevalence, such as Pakistan and some Canadian provinces.5Borhany, M., Fatima, H., Naz, A., Patel, H., & Shamsi, T., (2012). Pattern of bleeding and response to therapy in Glanzmann thrombasthenia. Haemophilia: the official journal of the World Federation of Hemophilia, 18(6), e423–e425. Lee, A., & Poon, M. C. (2018). Inherited platelet functional disorders: General principles and practical aspects of management. Transfusion and apheresis science: official journal of the World Apheresis Association: official journal of the European Society for Haemapheresis, 57(4), 494–501. 6(Krause, K. A., & Graham, B. C. (2023, August 28). Glanzmann thrombasthenia. In StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK538270/
Epidemiologists attribute this increase in cases to consanguineous marriages. For example, a recent study from Upper Egypt found that Glanzmann thrombasthenia was relatively more common there, likely due to the increased rate of consanguinity in the region.7 Abdel Hakeem Khalifa, G. L., El-Sayed, A. A., Elmasry, Z., Elsayh, K. I., Atwa, Z. T., Morgan, D. S., Hassan, E. E., Hassan, M. A., & Youssef, M. A. (2025). Epidemiological and clinical characteristics of children and young adults with Glanzmann’s thrombasthenia in Upper Egypt: A multicenter cross-sectional study. Annals of Hematology, 104(3), 1961.
What causes Glanzmann Thrombasthenia?
Integrin αIIbβ3 is a complex made up of two subunits: αIIb and β3. These subunits are encoded by genes ITGA2B and ITGB3, respectively.
These genes are located on chromosome 17 and expressed independently. Glanzmann Thrombasthenia is primarily caused by genetic mutations that affect the ITGA2B or ITGB3 genes. People either inherit these mutations from their parents or acquire them later in life.
Inherited Mutations:
Glanzmann Thrombasthenia is an autosomal recessive disorder. This means that an individual must inherit mutations in both copies (alleles) of the affected gene to develop the condition. If we talk about the outcomes of these mutations:
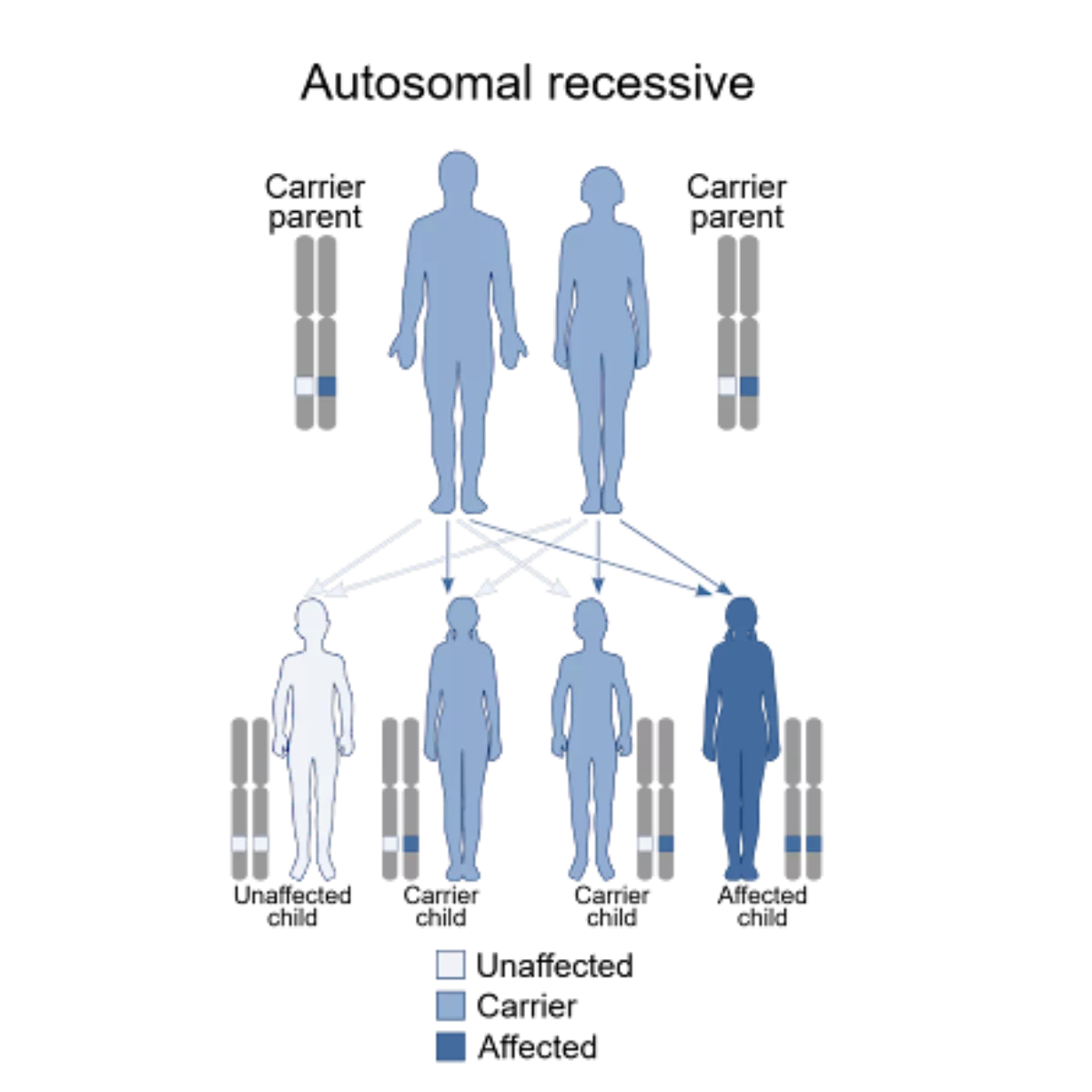
Image Courtesy: Wikimedia Commons, under CC license
- Some mutations impair the biosynthesis or transportation of subunits αIIb and β3, preventing their proper assembly and expression on the platelet surface.
- Other mutations alter the ligand-binding pocket or lock the integrin in a continuously activated state, disturbing normal platelet aggregation.
Homozygous vs Compound Heterozygous
The pattern of inheritance influences the severity and type of GT:
- Homozygous: In the homozygous form, identical mutations in both alleles (copies) of the same gene result in a completely absent αIIbβ3 receptor. This is typically seen in type I GT.
- Compound Heterozygous: Two different mutations in the same genes cause the partial synthesis of αIIbβ3 with variable receptor function. This genotype is present in type II and variant types of GT.(2)
Acquired Mutations:
Although rare, mutations can sometimes be acquired secondary to certain diseases, such as multiple myeloma8Solh, T., Botsford, A., & Solh, M., 2015). Glanzmann’s thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. Journal of Blood Medicine, 6, 219–227. and systemic lupus erythematosus (SLE) 9Nurden, A. T., Fiore, M., Nurden, P., & Pillois, X., 2011). Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood, 118(23), 5996–6005. . Scientists believe this happens because these diseases activate the immune system to produce autoantibodies such as the anti-alpha IIb beta3, which target the αIIbβ3 integrin receptor on platelets.
Symptoms of Glanzmann Thrombasthenia
Most patients of GT present in childhood with bleeding from mucosal surfaces and the skin. Statistics from registries and cohorts show that patients often report: 10Usman, M., Khan, M., Shahbaz, N., Zaffar, L., Tariq, H., Iftikhar, R., Ghafoor, T., Khan, M. A., & Zafar, T. (2024). Bleeding Phenotype of Glanzmann Thrombasthenia (GT) and Treatment Outcomes in Over One Hundred Patients: A Two-Center Experience in North Pakistan. Cureus, 16(11), e73724. https://doi.org/10.7759/cureus.73724
- Easing Bruising: Sometimes, after a minor trauma
- Nosebleeds (Epistaxis)
- Gum (Gingival) bleeding
- GIT bleeding manifesting as blood in vomiting, tarry black stools, or fresh blood per rectum
- Heavy or prolonged menstrual bleeding (Menorrhagia): In one systematic review, 22% of women with GT reported heavy menstrual bleeding. 11Punt, M. C., Schuitema, P. C. E., Bloemenkamp, K. W. M., Kremer Hovinga, I. C. L., & van Galen, K. P. M. (2020). Menstrual and obstetrical bleeding in women with inherited platelet receptor defects- A systematic review. Haemophilia: the official journal of the World Federation of Hemophilia, 26(2), 216–227. https://doi.org/10.1111/hae.13927
- Post-surgery or post-trauma bleeding: Severity varies from person to person. Some patients may bleed profusely from a minor injury, while others may just have bruising after a major trauma.
Complications of Glanzmann Thrombasthenia
The complications of Glanzmann Thrombasthenia include:
- Iron-deficiency anemia: Chronic bleeding in the gastrointestinal tract (GIT) may cause iron deficiency anemia, characterized by fatigue, weakness, and pallor.
- Obstetric risks: GT patients face a higher risk of bleeding during pregnancy, labor, and delivery. Repeated exposure to transfused platelets triggers the body to produce antibodies against them. These antibodies may cross the placenta and attack fetal or neonatal platelets, leading to thrombocytopenia (i.e, a decreased number of platelets).12Punt, M. C., Schuitema, P. C. E., Bloemenkamp, K. W. M., Kremer Hovinga, I. C. L., & van Galen, K. P. M. (2020). Menstrual and obstetrical bleeding in women with inherited platelet receptor defects- A systematic review. Haemophilia: the official journal of the World Federation of Hemophilia, 26(2), 216–227. https://doi.org/10.1111/hae.13927
- Intracranial bleeding: It’s a life-threatening condition that affects 1-2% of the total cases.13Abou-Ismail, M. Y., Diamond, A., Kaplan, M., Meeks, B., & Shapiro, A. D. (2020). Glanzmann thrombasthenia: Insights into treatment and ongoing clinical research. Haematologica, 105(12), 2738–2747. https://haematologica.org/article/view/9325
- GIT bleeding: A small mucosal injury can cause bleeding for hours in GT.
How to diagnose Glanzmann Thrombasthenia?
According to the Subcommittee on Platelet Physiology, part of the International Society on Thrombosis and Haemostasis (ISTH), the diagnosis of GT is based on a clear clinical history of bleeding disorders, a series of preliminary investigations, and then some confirmatory tests.14.Krause KA, Graham BC. Glanzmann Thrombasthenia. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538270/
Clinical Evaluation:
In the clinical history, there are episodes of uncontrolled or profuse bleeding following a major event such as circumcision, loss of the first milk tooth, or the first period. The majority of GT patients get diagnosed before the age of 14, based on a positive history for:
- Prolonged and frequent nosebleeds
- Heavy menstrual bleeding
- Easy bruising
In adults, doctors suspect GT often after an episode of ongoing bleeding following injuries or surgeries.
During a physical examination, your doctor will likely look for:
- Petechiae: tiny, pinpoint red or purple spots
- Echymosis: bruise-like area, larger than petechiae
- Any current bleeding
Preliminary Laboratory Tests:
Once your doctor suspects GT, they will run a couple of labs to rule out common bleeding disorders. These labs include:
- Complete Blood Count (CBC) to rule out thrombocytopenia. A patient of GT would have a normal platelet count in CBC.
- Coagulation Profile: This comprises prothrombin time (PT) and activated partial thromboplastin time (aPTT). If you have GT, these labs would also be within the normal range. But it would be deranged in the case of other bleeding disorders.
- Assessment for von Willebrand Disease: This includes tests such as von Willebrand Factor (vWF) antigen, ristocetin cofactor activity, and factor VIII coagulant activity, to rule out more common causes of uncontrolled bleeding.
- Peripheral Smear: It helps identify any abnormalities in the morphology of platelets. A normal morphology strengthens the suspicion of GT.
Screening Tests:
Though not specific to GT, these tests help screen for platelet functional abnormalities:
Platelet Function Analyzer or PFA-100
Even though it’s not a part of ISTH recommendations, many scientists consider it a useful screening test for Glanzmann Thrombasthenia. In this test, your blood sample is passed through a device, under shear stress, simulating our body’s internal environment. For a normal person, this would activate the platelets, causing them to aggregate and form a plug that would stop blood flow, resulting in a shorter bleeding time. On the other hand, since GT patients have a defective platelet aggregation, this test would yield a prolonged bleeding time with their samples.
Light Transmission Aggregometry (LTA)
LTA is a gold-standard test to assess platelet function. It involves evaluating how well light passes through a sample of platelet-rich plasma, in the presence of agonists such as epinephrine, ADP, or collagen.
First, they centrifuge your blood sample to get platelet-rich plasma (PRP). Then, they pass this PRP through an aggregometer. The aggregometer shines a beam of light through the sample, continuously measuring light transmission. This makes PRP cloudy as the suspended platelets scatter light and reduce its transmission. Then, they add the platelet agonists to see if the platelets aggregate and incur a change in light transmission. Now, if the patient is healthy, his platelets would clump together, allowing for more space for the light to pass, enhancing light transmission. But, if the patient has Glanzmann Thrombasthenia, platelet aggregation in response to all agonists, except ristocetin, would be impaired, leading to little or no change in light transmission.15Hayward, C. P., Pai, M., Liu, Y., Moffat, K. A., Seecharan, J., Webert, K. E., Cook, R. J., & Heddle, N. M. (2009). Diagnostic utility of light transmission platelet aggregometry: results from a prospective study of individuals referred for bleeding disorder assessments. Journal of thrombosis and haemostasis: JTH, 7(4), 676–684. https://doi.org/10.1111/j.1538-7836.2009.03273.x
Confirmatory Tests:
These investigations help establish the molecular and genetic basis for Glanzmann thrombasthenia:
Flow Cytometry
Flow cytometry is the confirmatory test for GT that assesses the expression of platelet surface glycoprotein (GPIIb/IIIa). During the test, platelets are labeled with monoclonal antibodies such as CD41 and CD61 for glycoproteins GPIIb and GPIIIa, respectively. These antibodies are also conjugated with fluorescent markers to help with visualization. Then the mean fluorescence intensity (MFI), which reflects the surface expression of αIIbβ3, is measured.16Saraymen, B., Muhtaroğlu, S., Köker, M. Y., Sarper, N., Zengin, E., Albayrak, C., Albayrak, D., Zülfikar, B., Koç Şenol, B., Bentli, E., Yılmaz, S., Çetin, A., Eser, B., & Çetin, M. (2021). Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of thirty-two Turkish patients with Glanzmann thrombasthenia: a multicenter experience. Turkish journal of medical sciences, 51(4), 2135–2141. https://doi.org/10.3906/sag-2006-107
This test not only helps diagnose GT but also identifies the respective subtype. For example:
- In Type I GT, there is expression of both glycoproteins CD41 and CD61 is reduced (<5% of the normal)
- Type II GT: Expression of CD41 and CD61 is detectable but significantly reduced, usually between 5-20% of the normal.
- Variant Type: Near-normal expression of CD41 and CD61 on flow cytometry, but the αIIbβ3 complex is functionally defective.
Genetic Testing or Next Generation Sequencing (NGS)
This method detects the mutations in the ITGA2B and ITGB3 genes, which code for the GPIIb (CD41) and GPIIIa (CD61) proteins. In families with an identified mutation, targeted genetic testing can help detect carriers and allow for prenatal diagnosis, particularly in consanguineous marriages.
Differential Diagnosis of Glanzmann Thrombasthenia (GT)
When evaluating a patient with mucocutaneous bleeding and a normal platelet count, healthcare providers should keep these differentials in mind:
Acquired Glanzmann Syndrome (Autoimmune):
- Autoantibodies against GPIIb/IIIa
- Associated with autoimmune diseases or malignancies
- Findings similar to congenital GT
- Normal receptor expression but impaired function
- No lifelong bleeding history
Bernard-Soulier Syndrome (BSS):
- Defect in GPIb-IX-V complex, instead of GPIIb/IIIa
- Reduced platelet count with the presence of abnormally large platelets.
- Absent aggregation with ristocetin
- Reduced GPIb expression on flow cytometry
Platelet Storage Pool Disorders:
- Deficiency of alpha or dense granules
- Defective release reaction, not receptor issue
- Variable platelet aggregation response
- Abnormal granules are seen on electron microscopy
Medication-Induced Platelet Dysfunction:
- Caused by aspirin, NSAIDs, clopidogrel, and SSRIs
- Inhibits normal platelet function
- History of recent drug use
- Improvement in symptoms upon withdrawing the causative agent
Is Glanzmann Thrombasthenia different from Von Willebrand Disease?
Even though Glanzmann Thrombasthenia (GT) and von Willebrand Disease (vWD) are inherited bleeding disorders and they share some clinical features, but they differ in many aspects, such as:
| Feature | Glanzmann Thrombasthenia (GT) | von Willebrand Disease (VWD) |
| Primary Hemostatic Defect | Impaired platelet aggregation due to qualitative or quantitative defects in the expression of integrin αIIbβ3. | Impaired platelet adhesion because of the deficiency or dysfunction of von Willebrand factor (VWF) |
| Genetic Inheritance | Autosomal recessive, caused by mutations in the ITGA2B or ITGB3 genes. | Type 1 and most Type 2 are autosomal dominant, while Type 3 and some Type 2 variants are autosomal recessive. |
| Platelet Aggregation Studies | Absent aggregation on LTA test with all agonists except ristocetin. | Normal aggregation with all agonists except ristocetin. |
| Laboratory Diagnosis | Normal platelet count with abnormal platelet aggregation profile. Flow cytometry shows absent or reduced αIIbβ3 expression, which is confirmed by molecular genetic testing. | Low VWF antigen and/or ristocetin cofactor activity. Multimer analysis for VWF subtype classification, confirmed by VWF gene sequencing. |
| Treatment Options | Antifibrinolytics, platelet transfusion, recombinant factor VIIa (rFVIIa), and hematopoietic stem cell transplantation (in refractory or severe cases). | Desmopressin (DDAVP) for Type 1 and mild Type 2, and plasma-derived VWF/FVIII concentrates for severe bleeding or Type 3.Antifibrinolytics and hormonal control for menorrhagia. |
| Prevalence | Extremely rare (~1 in 1,000,000); higher incidence in consanguineous populations | The most common inherited bleeding disorder (1% prevalence17Data and Statistics on von Willebrand Disease. (2024, May 15). Von Willebrand Disease (VWD). https://www.cdc.gov/von-willebrand/data/index.html), often underdiagnosed due to mild symptoms. |
| Gender Distribution | Equal frequency in males and females. | Affects both sexes, but bleeding tends to be more clinically evident in females due to menstruation and childbirth. |
Treatment of Glanzmann Thrombasthenia
GT is a bleeding disorder, and its treatment revolves around managing bleeding through a combination of supportive measures, pharmacotherapy and transfusions.
For Mild to Moderate bleeding:
- The first line of treatment is local hemostatic measures such as topical thrombin, ice therapy, or compression, as seen with nasal packing in cases of epistaxis.
- Antifibrinolytics such as traxemic acid or amino-caproic acid may be combined with local hemostatic measures, especially for oral, nasal, or menstrual bleeding.
For Moderate to Severe bleeding or Surgical Prophylaxis:
For moderate to severe bleeding or surgical prophylaxis platelet transfusion is the mainstay of treatment. Your healthcare provider will often ask for it when you are actively bleeding or are about to undergo surgery.
Since the number of platelets is normal in GT, blood CP is of little to no value in assessing how effective the transfusion is. So, your doctor will clinically analyze the effectiveness of treatment through an improvement or resolution of your bleeding.
For Heavy Menstrual Bleeding:
Your gynaecologist may consider hormonal therapy like combined oral contraceptives or progestins. These help regulate the cycle and reduce bleeding in females with GT.
Hematopoietic Stem Cell Transplantation:
Also known as bone marrow transplant, it is the only cure for GT so far. Your doctor might advise it when no other treatment works. In this procedure, healthy stem cells from a donor are infused into the patient. These stem cells then differentiate into healthy blood cells, including platelets, yielding a normal clotting mechanism.
Gene Therapy:
It involves correcting the underlying genetic defect by introducing a functional copy of the affected gene into the patient’s stem cells. Although the early results are promising, it’s still in the experimental phase.18Wilcox, D. A., & White, G. C., 2nd (2003). Gene therapy for platelet disorders: studies with Glanzmann’s thrombasthenia. Journal of thrombosis and haemostasis : JTH, 1(11), 2300–2311.
Treatment Complications:
Patients with GT require lifelong transfusions. Due to repeated transfusions, you may have antibodies produced against the platelets of the donor.19Gabe, C., Ziza, K. C., Durazzo, N., Pagani, F. M., Oliveira, V. B., Conrado, C., Dezan, M. R., Alfredo Mendrone, J., Villaça, P. R., Dinardo, C. L., & Rocha, V. (2022). Detection of alloimmunization in Glanzmann Thrombasthenia and Bernard-Soulier Syndrome: Data from a Brazilian Center. Hematology, Transfusion and Cell Therapy, 45(Suppl 2), S101. https://doi.org/10.1016/j.htct.2022.06.005 This is called alloimmunization. Over time, these antibodies destroy the incoming platelets, making future transfusions less effective. This condition is called platelet refractoriness.
- To prevent these conditions, doctors use blood products that are deficient in leukocytes.
- If at all, alloimmunization or platelet refractoriness develops, recombinant activated factor VII (rFVIIa) becomes the treatment of choice. rFVIIa bypasses the platelet aggregation pathway by directly promoting thrombin generation and fibrin formation.
Patient Counselling:
As with any other chronic illness, lifestyle advice and patient counselling are essential in Glanzmann Thrombasthenia. Healthcare providers need to educate their patients and caregivers in the following areas:
- Avoid contact sports. Even minor injuries can cause serious bleeding.
- Use soft toothbrushes and avoid flossing harshly to minimize gum bleeding.
- Avoid drugs (like NSAIDs, aspirin) that worsen your bleeding. For pain or fever, you can use paracetamol – it’s safer.
- Carry a medical alert ID and keep emergency contact information readily available at all times.
- For females with menorrhagia, discuss early use of hormonal therapy and monitor for iron deficiency.
- Inform all healthcare providers (including dentists) of the diagnosis before any procedure.
- For emotional and long-term support, consider referral to patient support groups or genetic counselling, especially in family planning discussions.
Life Expectancy in Glanzmann Thrombasthenia
Glanzmann Thrombasthenia is not a life-shortening illness. With modern medical care, the majority of patients have a normal life expectancy with a good prognosis. Deaths are rare and are linked to severe uncontrolled bleeding, complications from transfusion, or lack of access to care. So, if you have access to good medical care that is prepared for emergencies, follow your doctor’s advice religiously, and make smart lifestyle choices, you can have a full, active life, pursue your career, and even manage menstruation and pregnancy successfully.
Conclusion
Glanzmann thrombasthenia is a rare genetic platelet function disorder characterized by lifelong mucocutaneous bleeding that affects males and females equally. Diagnosis is based on clinical evaluation and specialized tests like Light Transmission Aggregometry and Flow Cytometry. Treatment follows a tiered approach, depending upon the severity of blood loss. With early diagnosis, timely treatment and proper lifestyle modification, the majority patients lead normal lives.
Refrences
- 1Nurden A. (2021). Profiling the Genetic and Molecular Characteristics of Glanzmann Thrombasthenia: Can It Guide Current and Future Therapies?. Journal of blood medicine, 12, 581–599. https://doi.org/10.2147/JBM.S273053
- 2Botero, J. P., Lee, K., Branchford, B. R., Bray, P. F., Freson, K., Lambert, M. P., Luo, M., Mohan, S., Ross, J. E., Bergmeier, W., & Di Paola, J. (2020). Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica, 105(4), 888–894. https://doi.org/10.3324/haematol.2018.214239
- 3Nurden A. (2021). Profiling the Genetic and Molecular Characteristics of Glanzmann Thrombasthenia: Can It Guide Current and Future Therapies?. Journal of blood medicine, 12, 581–599. https://doi.org/10.2147/JBM.S273053
- 4Botero, J. P., Lee, K., Branchford, B. R., Bray, P. F., Freson, K., Lambert, M. P., Luo, M., Mohan, S., Ross, J. E., Bergmeier, W., & Di Paola, J. (2020). Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica, 105(4), 888–894. https://doi.org/10.3324/haematol.2018.214239
- 5Borhany, M., Fatima, H., Naz, A., Patel, H., & Shamsi, T., (2012). Pattern of bleeding and response to therapy in Glanzmann thrombasthenia. Haemophilia: the official journal of the World Federation of Hemophilia, 18(6), e423–e425. Lee, A., & Poon, M. C. (2018). Inherited platelet functional disorders: General principles and practical aspects of management. Transfusion and apheresis science: official journal of the World Apheresis Association: official journal of the European Society for Haemapheresis, 57(4), 494–501.
- 6
- 7Abdel Hakeem Khalifa, G. L., El-Sayed, A. A., Elmasry, Z., Elsayh, K. I., Atwa, Z. T., Morgan, D. S., Hassan, E. E., Hassan, M. A., & Youssef, M. A. (2025). Epidemiological and clinical characteristics of children and young adults with Glanzmann’s thrombasthenia in Upper Egypt: A multicenter cross-sectional study. Annals of Hematology, 104(3), 1961.
- 8Solh, T., Botsford, A., & Solh, M., 2015). Glanzmann’s thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. Journal of Blood Medicine, 6, 219–227.
- 9Nurden, A. T., Fiore, M., Nurden, P., & Pillois, X., 2011). Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood, 118(23), 5996–6005.
- 10Usman, M., Khan, M., Shahbaz, N., Zaffar, L., Tariq, H., Iftikhar, R., Ghafoor, T., Khan, M. A., & Zafar, T. (2024). Bleeding Phenotype of Glanzmann Thrombasthenia (GT) and Treatment Outcomes in Over One Hundred Patients: A Two-Center Experience in North Pakistan. Cureus, 16(11), e73724. https://doi.org/10.7759/cureus.73724
- 11Punt, M. C., Schuitema, P. C. E., Bloemenkamp, K. W. M., Kremer Hovinga, I. C. L., & van Galen, K. P. M. (2020). Menstrual and obstetrical bleeding in women with inherited platelet receptor defects- A systematic review. Haemophilia: the official journal of the World Federation of Hemophilia, 26(2), 216–227. https://doi.org/10.1111/hae.13927
- 12Punt, M. C., Schuitema, P. C. E., Bloemenkamp, K. W. M., Kremer Hovinga, I. C. L., & van Galen, K. P. M. (2020). Menstrual and obstetrical bleeding in women with inherited platelet receptor defects- A systematic review. Haemophilia: the official journal of the World Federation of Hemophilia, 26(2), 216–227. https://doi.org/10.1111/hae.13927
- 13Abou-Ismail, M. Y., Diamond, A., Kaplan, M., Meeks, B., & Shapiro, A. D. (2020). Glanzmann thrombasthenia: Insights into treatment and ongoing clinical research. Haematologica, 105(12), 2738–2747. https://haematologica.org/article/view/9325
- 14.Krause KA, Graham BC. Glanzmann Thrombasthenia. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538270/
- 15Hayward, C. P., Pai, M., Liu, Y., Moffat, K. A., Seecharan, J., Webert, K. E., Cook, R. J., & Heddle, N. M. (2009). Diagnostic utility of light transmission platelet aggregometry: results from a prospective study of individuals referred for bleeding disorder assessments. Journal of thrombosis and haemostasis: JTH, 7(4), 676–684. https://doi.org/10.1111/j.1538-7836.2009.03273.x
- 16Saraymen, B., Muhtaroğlu, S., Köker, M. Y., Sarper, N., Zengin, E., Albayrak, C., Albayrak, D., Zülfikar, B., Koç Şenol, B., Bentli, E., Yılmaz, S., Çetin, A., Eser, B., & Çetin, M. (2021). Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of thirty-two Turkish patients with Glanzmann thrombasthenia: a multicenter experience. Turkish journal of medical sciences, 51(4), 2135–2141. https://doi.org/10.3906/sag-2006-107
- 17Data and Statistics on von Willebrand Disease. (2024, May 15). Von Willebrand Disease (VWD). https://www.cdc.gov/von-willebrand/data/index.html
- 18Wilcox, D. A., & White, G. C., 2nd (2003). Gene therapy for platelet disorders: studies with Glanzmann’s thrombasthenia. Journal of thrombosis and haemostasis : JTH, 1(11), 2300–2311.
- 19Gabe, C., Ziza, K. C., Durazzo, N., Pagani, F. M., Oliveira, V. B., Conrado, C., Dezan, M. R., Alfredo Mendrone, J., Villaça, P. R., Dinardo, C. L., & Rocha, V. (2022). Detection of alloimmunization in Glanzmann Thrombasthenia and Bernard-Soulier Syndrome: Data from a Brazilian Center. Hematology, Transfusion and Cell Therapy, 45(Suppl 2), S101. https://doi.org/10.1016/j.htct.2022.06.005



