
Hunter syndrome (also known as mucopolysaccharidosis II or MPS II) is a rare X-linked recessive lysosomal storage disorder with a significantly higher prevalence in males. The estimated incidence is approximately 1 in 162,000 live male births.1Hashmi MS, Gupta V. Mucopolysaccharidosis Type II. [Updated 2023 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560829/ It is caused by a deficiency of lysosomal enzymes responsible for the degradation of mucopolysaccharides or glycosaminoglycans (GAGs), which results in abnormal accumulation of GAGs within lysosomes, leading to impaired cellular function and progressive damage to multiple organs and tissues. In easier words, it is the failure of the body’s ability to break down sugar molecules properly. This accumulation leads to a variety of clinical manifestations, affecting multiple organs such as the skin, musculoskeletal, respiratory, and cardiovascular systems.
Hunter Syndrome Causes
The root cause of Hunter syndrome is a deficiency of a specific lysosomal enzyme called iduronate-2-sulfatase (I2S). This absence of the enzyme leads to the accumulation of heparan sulfate and dermatan sulfate in various body tissues. Notably, Hunter syndrome is the only type of mucopolysaccharidosis (MPS) to demonstrate X-linked recessive inheritance.
Phenotypes of Hunter Syndrome:
There are two primary phenotypes of Hunter syndrome: severe and mild (also known as attenuated). The severe form is characterized by significant cognitive decline and behavioral issues, whereas the mild form involves little to no cognitive impairment. The severity of symptoms varies widely between individuals, but early diagnosis is crucial for effective management and treatment.
Hunter Syndrome Signs & Symptoms
The signs and symptoms of Hunter syndrome vary in presentation and involve multiple organs and tissues, which makes the diagnosis a challenge. Newborns do not show any signs or symptoms at the time of birth. As the children grow, the buildup of GAGs continues throughout the cells of the body, making signs of MPS II become more visible, and symptoms usually start to show when the child is between 18 months and four years of age.
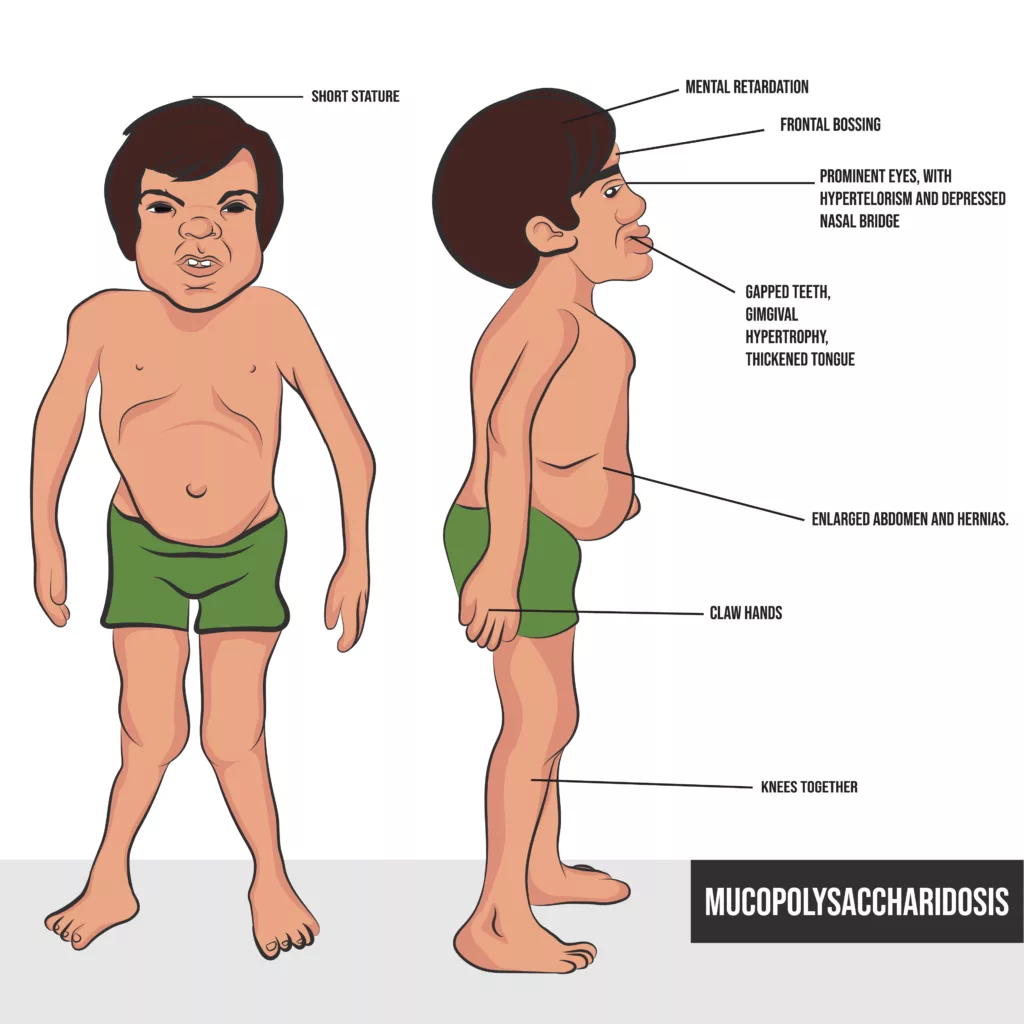
A child with Hunter syndrome might present with:
Neurological Features:
- Cognitive impairment, with difficulties in thinking, learning, and communication.
- Behavioral problems such as aggression, hyperactivity, or difficulty concentrating.
- Hydrocephalus, often seen as macrocephaly (enlarged head circumference).
Facial and Skeletal Characteristics:
- Coarse facial features, including a broad nose, thick lips, and a large tongue.
- Short stature and joint stiffness, leading to restricted movement.
Respiratory Issues:
- Frequent upper respiratory infections and chronic ear infections (otitis media).
- Obstructive airway complications and sleep apnea.
Abdominal and Organ Enlargement:
- Enlarged liver and spleen (hepatosplenomegaly).
- Abdominal hernias, including umbilical and inguinal hernias.
Cardiovascular Complications:
- Heart valve abnormalities that lead to cardiovascular dysfunction over time.
Oral and Dental Issues:
- Abnormal tooth development, malocclusion, jaw defects, and enamel malformation.
- Oral lesions and a thickened tongue that contribute to feeding or speech difficulties.
Skin Lesions:
- Pebbled, ivory-colored skin lesions are commonly found on the back, legs, and arms. These lesions are considered a hallmark of Hunter syndrome.
Hearing and Vision Problems:
- Progressive hearing loss and recurrent ear infections.
- Corneal clouding may occur in some cases, affecting vision.
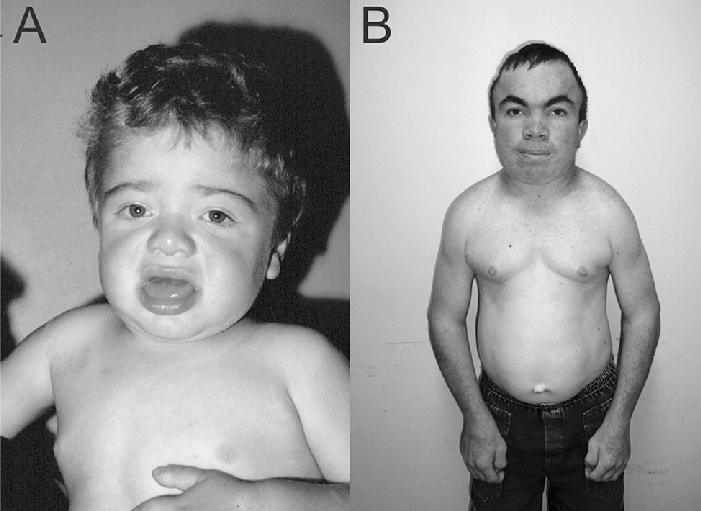
Hunter Syndrome Diagnosis
Due to the multisystem involvement in this syndrome, there is usually a need for extensive examination and laboratory testing to rule out other diseases with similar manifestations.
The diagnosis involves a combination of clinical assessments, enzyme activity assays, and genetic testing.
The following tests are carried out for the diagnosis:
Urine & Plasma GAG Levels:
This is usually the test conducted initially to screen individuals for this disease.2Stapleton, M., Kubaski, F., Mason, R. W., Yabe, H., Suzuki, Y., Orii, K. E., Orii, T., & Tomatsu, S. (2017). Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert opinion on orphan drugs, 5(4), 295–307. https://doi.org/10.1080/21678707.2017.1296761 The elevated plasma levels indicate the incidence of the disease, while the urinary GAG levels have lesser significance.3Giugliani, R., Villarreal, M. L., Valdez, C. A., Hawilou, A. M., Guelbert, N., Garzón, L. N., Martins, A. M., Acosta, A., Cabello, J. F., Lemes, A., Santos, M. L., & Amartino, H. (2014). Guidelines for diagnosis and treatment of Hunter Syndrome for clinicians in Latin America. Genetics and molecular biology, 37(2), 315–329. https://doi.org/10.1590/s1415-47572014000300003
Iduronate Sulfatase (IDS) Levels:
This test is considered the gold standard.4Neufeld, E. F., & Fratantoni, J. C. (1970). Inborn errors of mucopolysaccharide metabolism. Science (New York, N.Y.), 169(3941), 141–146. https://doi.org/10.1126/science.169.3941.141 Most cases with high severity have a very low amount of this enzyme.
Genetic Testing:
Molecular genetic testing is considered to be a confirmatory test. The confirmation is done by measuring I2S activity in fibroblast/leucocytes and analyzing I2S gene mutations.5Sestito, S., Falvo, F., Scozzafava, C., Apa, R., Pensabene, L., Bonapace, G., Moricca, M. T., & Concolino, D. (2018). Genetics and Gene Therapy in Hunter Disease. Current gene therapy, 18(2), 90–95. https://doi.org/10.2174/1566523218666180404155759
Imaging:
Radiographic X-rays with characteristic findings of lipid deposition in the skull, joints, axial skeleton, and limbs are known as dysostosis multiplex.6Stapleton, M., Kubaski, F., Mason, R. W., Yabe, H., Suzuki, Y., Orii, K. E., Orii, T., & Tomatsu, S. (2017). Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert opinion on orphan drugs, 5(4), 295–307. https://doi.org/10.1080/21678707.2017.1296761 Some of the characteristic findings include dwarfism, J-shaped lesions in sella turcica, scoliosis, kyphosis, hypoplastic thickened carpal, and tarsal bones.
Hunter Syndrome Treatment
Currently, there is no curative therapy for Hunter syndrome. Treatment plans require interdisciplinary coordination and depend greatly on the age of the child and the presentation of the specific disease. Research has led to the development of enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), and other supportive treatments, such as the usage of NSAIDs.
Enzyme Replacement Therapy (ERT):
Idursulfase (recombinant I2S) enzyme replacement therapy (ERT) addresses the root cause, the underlying enzyme deficiency, of this syndrome. It is an effective treatment as it improves walking ability and respiratory function and aids in reducing the size of the spleen and liver as well as the urinary glycosaminoglycan levels.7Daniele, A., Tomanin, R., Villani, G. R., Zacchello, F., Scarpa, M., & Di Natale, P. (2002). Uptake of recombinant iduronate-2-sulfatase into neuronal and glial cells in vitro. Biochimica et biophysica acta, 1588(3), 203–209. https://doi.org/10.1016/s0925-4439(02)00166-7 To achieve the best results with enzyme replacement therapy, it should be started before age six.8Grant, N., Sohn, Y. B., Ellinwood, N. M., Okenfuss, E., Mendelsohn, B. A., Lynch, L. E., Braunlin, E. A., Harmatz, P. R., & Eisengart, J. B. (2022). Timing is everything: Clinical courses of Hunter syndrome associated with age at initiation of therapy in a sibling pair. Molecular genetics and metabolism reports, 30, 100845. https://doi.org/10.1016/j.ymgmr.2022.100845
Hematopoietic Stem Cell Transplantation (HSCT):
Doctors consider hematopoietic stem cell transplantation (HSCT) to be a more effective therapy compared to enzyme replacement therapy (ERT). It is more cost-effective, as it is a one-time procedure. It has shown improvement in neurocognitive functions and decreases the rate of neurodegeneration seen by brain imaging. Ideally, doctors should perform HSCT on an affected child before neurological symptoms appear.
NSAIDs Usage:
The accumulation of GAGs in articular cartilage, extracellular matrix, and tendons at joints usually initiates an inflammatory cascade, which causes erosive dysplasia and degenerative changes in the articular cartilage. Non-steroidal anti-inflammatory drugs (NSAIDs) usage inhibits the production of inflammatory mediators and prevents the associated degenerative joint changes.
In managing Hunter syndrome, additional interventions should address the multi-organ manifestations, including surgical repair of abdominal/inguinal hernias and cardiac valve replacements.
Hunter Syndrome Prognosis
While early detection and appropriate and timely management may improve the quality of life, the truth is that the prognosis of Hunter Syndrome is poor. The outcome is worse for the severe phenotype than the mild form. Patients with severe form have a life expectancy of 10 to 20 years, and those with mild form usually live anywhere between 20 and 60 years.
The majority of the patients die by the second decade of life due to pulmonary dysfunction, cardiac valvular abnormalities, or a combination of both.
Do children with Hunter syndrome live normal lives?
Some children with less severe Hunter syndrome grow up and live long lives. They will achieve puberty like normal teenagers and can have children. But heart disease and trouble breathing can still cause problems for them. Early intervention can help children with even severe Hunter syndrome participate in play, build friendships, and enjoy activities similar to other children despite their physical differences.
Can females also be affected by Hunter Syndrome?
Although primarily affecting males, females can carry one altered copy of the I2S gene without showing symptoms. Due to having two X chromosomes, females only develop MPS 2 if both carry the defective gene. This rarity explains why Hunter syndrome is very uncommon in females despite documented cases.
Prevention of Hunter Syndrome
Carrying a defective gene for Hunter syndrome makes complete prevention impossible. However, early intervention can help prevent some long-term complications. Enzyme replacement therapy (ERT) can help slow the progression of the disease for boys with milder Hunter syndrome by replacing the protein structure that their body is unable to make.
Hunter Syndrome Vs. Hurler Syndrome
Both of these syndromes share striking similarities in their clinical signs and symptoms, such as coarse facial features and respiratory, musculoskeletal, and cardiovascular manifestations. However, they differ in their underlying cause (etiology).
- Hurler syndrome arises from a mutation in the IDUA gene. This mutation can occur spontaneously, but there is an increased risk for parents with a family history of MPS I to have a child with the condition.9Sakuru R, Bollu PC. Hurler Syndrome. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532261/
- Hurler syndrome impacts males and females equally and is considered a rare disorder, with roughly 1 in every 100,000 babies born with the condition. We can distinguish Hunter syndrome from Hurler syndrome by several factors: X-linked recessive inheritance, slower disease progression, longer lifespan, and lack of corneal clouding.
- Diagnosis and treatment for Hurler’s syndrome involves the same approach as Hunter syndrome: Enzyme replacement therapy(ERT), hematopoietic stem cell transplant (HSCT), and symptomatic treatments depending on the organ involved.
Conclusion
This condition presents a formidable challenge to those affected, their families, and their medical caregivers.
As research continues, advancements in understanding the disorder and developing innovative treatments offer hope for improved outcomes. With each piece of research, we move towards improving the quality of life of these affected individuals.
Refrences
- 1Hashmi MS, Gupta V. Mucopolysaccharidosis Type II. [Updated 2023 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560829/
- 2Stapleton, M., Kubaski, F., Mason, R. W., Yabe, H., Suzuki, Y., Orii, K. E., Orii, T., & Tomatsu, S. (2017). Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert opinion on orphan drugs, 5(4), 295–307. https://doi.org/10.1080/21678707.2017.1296761
- 3Giugliani, R., Villarreal, M. L., Valdez, C. A., Hawilou, A. M., Guelbert, N., Garzón, L. N., Martins, A. M., Acosta, A., Cabello, J. F., Lemes, A., Santos, M. L., & Amartino, H. (2014). Guidelines for diagnosis and treatment of Hunter Syndrome for clinicians in Latin America. Genetics and molecular biology, 37(2), 315–329. https://doi.org/10.1590/s1415-47572014000300003
- 4Neufeld, E. F., & Fratantoni, J. C. (1970). Inborn errors of mucopolysaccharide metabolism. Science (New York, N.Y.), 169(3941), 141–146. https://doi.org/10.1126/science.169.3941.141
- 5Sestito, S., Falvo, F., Scozzafava, C., Apa, R., Pensabene, L., Bonapace, G., Moricca, M. T., & Concolino, D. (2018). Genetics and Gene Therapy in Hunter Disease. Current gene therapy, 18(2), 90–95. https://doi.org/10.2174/1566523218666180404155759
- 6Stapleton, M., Kubaski, F., Mason, R. W., Yabe, H., Suzuki, Y., Orii, K. E., Orii, T., & Tomatsu, S. (2017). Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert opinion on orphan drugs, 5(4), 295–307. https://doi.org/10.1080/21678707.2017.1296761
- 7Daniele, A., Tomanin, R., Villani, G. R., Zacchello, F., Scarpa, M., & Di Natale, P. (2002). Uptake of recombinant iduronate-2-sulfatase into neuronal and glial cells in vitro. Biochimica et biophysica acta, 1588(3), 203–209. https://doi.org/10.1016/s0925-4439(02)00166-7
- 8Grant, N., Sohn, Y. B., Ellinwood, N. M., Okenfuss, E., Mendelsohn, B. A., Lynch, L. E., Braunlin, E. A., Harmatz, P. R., & Eisengart, J. B. (2022). Timing is everything: Clinical courses of Hunter syndrome associated with age at initiation of therapy in a sibling pair. Molecular genetics and metabolism reports, 30, 100845. https://doi.org/10.1016/j.ymgmr.2022.100845
- 9Sakuru R, Bollu PC. Hurler Syndrome. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532261/



