
Xeroderma pigmentosum (XP) is a rare inherited disorder that causes extreme sensitivity to ultraviolet (UV) radiation from sunlight. It severely impairs the skin’s ability to repair DNA damage caused by UV exposure, dramatically increasing the risk of skin cancer and other complications..
What is Xeroderma Pigmentosa?
“Xero” means dry, and “derma” refers to skin, while “pigmentosum” highlights the grayish-brown pigmentation that often develops in affected individuals. XP is a rare autosomal recessive genodermatosis caused by defects in the nucleotide excision repair (NER) genes responsible for fixing UV-induced DNA damage.
Under normal circumstances, the body repairs damaged DNA, but in XP, UV radiation directly impairs the DNA repair mechanism. As a result, DNA damage caused by sunlight accumulates, leading to early skin aging and a much higher risk of skin cancers.
People with XP are extremely sensitive to the sun’s ultraviolet (UV) rays—both UVA and UVB. Even minimal UV exposure can lead to severe sunburns and blistering, sometimes as early as infancy.1Black J. O. (2016). Xeroderma Pigmentosum. Head and neck pathology, 10(2), 139–144. https://doi.org/10.1007/s12105-016-0707-8
This heightened sensitivity often causes early freckling and areas of lighter or uneven pigmentation. The skin may also become excessively dry, which is one of the more visible features of the condition.
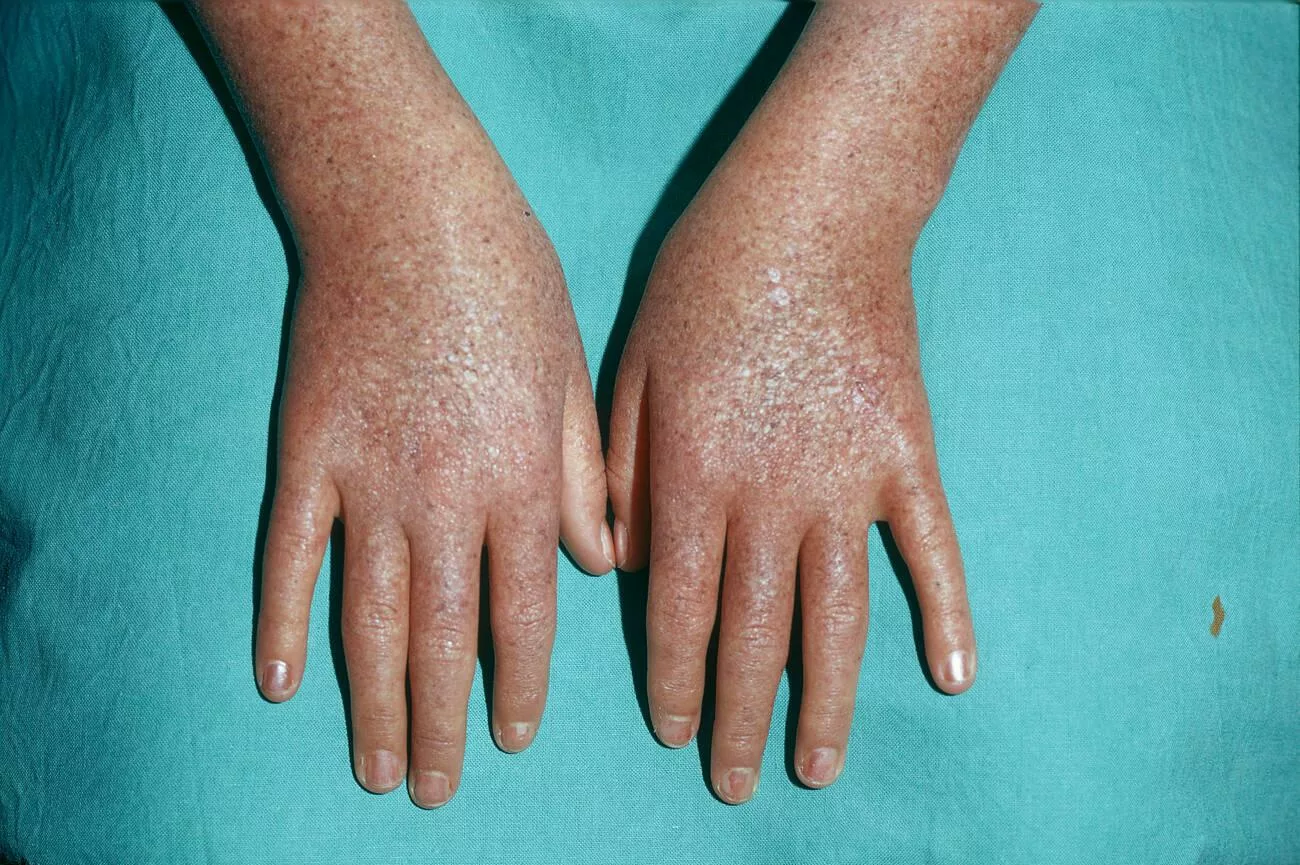
Clinical Manifestations of Xeroderma Pigmentosa:
The symptoms and indications of Xeroderma Pigmentosa usually appear in infancy or as soon as infants are exposed to sunlight. Mostly, the patients encounter the following symptoms.
- Burning of Skin: Regardless of skin color, patients with XP experience an intense burning sensation after even minimal sun exposure. This can begin in infancy and often results in blistering and redness.
- Freckling: Freckles appear as early as two years old in sites that are exposed to light. Patients mostly have wrinkling faces, necks, hands, feet, and usually bare skin.
- Solar Keratosis: Actinic (solar) keratoses, brown-coloured skin outgrowths on the skin, are common in patients of Xeroderma pigmentosum.
- Photophobia: XP patients often develop photophobia (light sensitivity), not fear of light, but discomfort or pain in bright light due to ocular involvement.
- Skin cancers: There is a probability of developing skin cancers, which are usually squamous cell carcinoma, basal cell carcinoma, or melanoma. The probability of developing skin cancers on the face, eyes, and tip of the lips is high.
- Xerosis: Presentation of extremely dry Skin is seen in patients, as indicated in the name of the disease. This dry Skin aggravates the overall symptoms of the disease and adds to the itching and burning sensations of patients.
Pathological changes in Skin:
- Those with xeroderma pigmentosum are born with normal Skin, but pigmentary changes begin to appear as they age and spend more time in the sun.
- The face, neck, chest, dorsal hands, and forearms of patients frequently exhibit hyperpigmented and hypopigmented macules.
- A scaly to hyperkeratotic erythematous macule or papule is the usual presentation of squamous cell carcinomas (SCC). These non-melanoma skin malignancies could exhibit a non-healing or easily traumatized wound as their initial symptom.
These non-melanoma skin tumors may seem like a wound that doesn’t heal or is vulnerable to damage. - Additionally, patients may exhibit skin malignancies like keratocanthomas. Keratoacanthomas may appear rapidly as dome-shaped nodules with central keratin plugs. These are considered a variant of SCC.
- Facial and oral malignant tumors, particularly those of the anterior tongue, can also be present in xeroderma pigmentosum patients.
Ocular Manifestations:
Multiple ophthalmologic signs of the disease are frequently seen in individuals with xeroderma pigmentosum.
- Patients often complain of progressively worsening eye issues over time.
- There may be indications of dry eyes or photophobia in the patients. Hyperpigmentation of the conjunctiva and eyelids is a potential complication of XP.
- Ectropion, corneal vascularization, or opacification are other symptoms that XP patients may exhibit. Additionally, patients may come with benign lid papillomas, basal cell carcinomas of the eyelids, or malignant melanoma, among other ocular and periocular lesions that can be either benign or cancerous.
Neurological Manifestation:
Neurological symptoms are progressive over time, regardless of sun exposure.
- During childhood, more symptoms may start to emerge. These mainly consist of weakened, thin skin with scarring spider veins (telangiectasia)
- Hearing loss, memory loss, and learning difficulties are all neurological or neurologically related conditions.
- Secondary to neuronal loss, cortical atrophy, and ventricular enlargement are neurologic symptoms.
- Sensorineural deafness and delayed neurodevelopment are hallmark features in patients with neurological involvement. Not all patients develop neurological symptoms, but those who do often experience a gradual worsening over time.
What causes Xeroderma Pigmentosum?
Genetic Inheritance:
As discussed before, Xeroderma pigmentosum is caused by inherited mutations in genes responsible for repairing UV-induced DNA damage, particularly within the nucleotide excision repair (NER) pathway. It follows an autosomal recessive pattern in most cases.2Black J. O. (2016). Xeroderma Pigmentosum. Head and neck pathology, 10(2), 139–144. https://doi.org/10.1007/s12105-016-0707-8
The hallmark of XP is the body’s inability to repair UV-damaged DNA, which leads to the accumulation of mutations, especially in skin cells. This failure underlies the extreme UV sensitivity, frequent sunburns, and increased skin cancer risk seen in all XP types
Extreme sensitivity to sunlight is always present, regardless of the genes involved, which differ from type to type. If the skin isn’t protected, it will inevitably result in painful sunburns and, subsequently, skin cancer.
Pattern of Inheritance of Disease:
There are eight identified subtypes of XP (XP-A to XP-G and XP-V). Seven of these are inherited in an autosomal recessive manner. This means both parents typically carry one defective copy of the gene but do not show symptoms themselves.
There is no well-established autosomal dominant form of classic XP. However, a few rare DNA repair disorders may share overlapping features but follow different inheritance patterns. So, the statement that “XP dominant type” exists is not accurate based on the current scientific classification.3Lehmann, A. R., McGibbon, D., & Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet Journal of Rare Diseases, 6(70). https://doi.org/10.1186/1750-1172-6-70
Risk factors for Xeroderma Pigmentosum
As it is a genetic disorder, certain factors increase the risk of inheriting or passing it on:
- Having a parent diagnosed with XP increases the chance of inheritance if both parents are carriers.
- If both parents are asymptomatic (silent) carriers of a mutated nucleotide excision repair gene, there is a 25% chance their child will inherit the condition.4Lehmann, A. R., McGibbon, D., & Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet Journal of Rare Diseases, 6(70). https://doi.org/10.1186/1750-1172-6-70
- Being a carrier (heterozygous for the XP gene mutation) typically does not cause symptoms but still allows the gene to be passed to offspring.
While not a genetic risk factor, smoking may worsen skin damage in diagnosed XP patients by increasing oxidative stress and compromising skin repair, making UV-related damage even more severe.5DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426
Diagnosis of Xeroderma Pigmentosum
Early diagnosis is crucial for XP management and can begin with genetic counseling for families with a known history of the condition, even as early as pregnancy.
Screening Options for XP:
For those with XP who are known or suspected, current recommendations for screening include:6Bradford, P. T., Goldstein, A. M., Tamura, D., Khan, S. G., Ueda, T., Boyle, J., … & Kraemer, K. H. (2011). Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. Journal of Medical Genetics, 48(3), 168–176. https://doi.org/10.1136/jmg.2010.082511
- Full-body skin exams every 3 to 6 months by a dermatologist experienced in skin cancer surveillance.
- Regular eye examinations by an ophthalmologist to check for UV-related damage to the cornea and retina.
- Dental assessments to monitor for oral lesions and mucosal involvement.
- Standard neurologic examination as advised by your primary healthcare provider.
Prenatal Diagnosis
Amniocentesis is a common invasive fetal diagnostic procedure that can be performed if you’re expecting and want to know your fetus’s risk of XP. This examination examines the fetal fluid. It can reveal whether the fetus carries the XP gene or other genetic disorders.7Piccione, M., Belloni Fortina, A., Ferri, G., Andolina, G., Beretta, L., Cividini, A., De Marni, E., Caroppo, F., Citernesi, U., & Di Liddo, R. (2021). Xeroderma Pigmentosum: General Aspects and Management. Journal of personalized medicine, 11(11), 1146. https://doi.org/10.3390/jpm111111468Kleijer, W. J., Laugel, V., Berneburg, M., Nardo, T., Fawcett, H., Gratchev, A., … & Lehmann, A. R. (2008). Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair, 7(5), 744–750. https://doi.org/10.1016/j.dnarep.2008.01.004
Management of Xeroderma Pigmentosa
Efficient management of Xeroderma pigmentosa includes early diagnosis and prompt treatment.
Treatment of XP:
Since it is a genetic disorder, Xeroderma pigmentosa has no definitive treatment to cure it completely. However, early intervention and strict sun protection can significantly reduce complications.
- Strict avoidance and protection from the sun is the most efficient way to lower the number of malignant tumors in XP patients. The best ways to limit UV radiation exposure should be made clear to patients and their caregivers.
- Patients should not leave their residences during the day.
- One should apply broad-spectrum sunscreen (SPF 50 or higher) every two hours. Along with sunscreen, patients should use lip balm.
- Patients should wear long sleeves and trousers as sun protection.
- Additionally, they want to put on a cap and side-shielded sunglasses.
- There should be a UV-blocking film on all windows in the house, automobile, and school.
- Avoid indoor UV sources such as fluorescent, halogen, and metal halide lights that emit UV radiation.9Kraemer, K. H., Patronas, N. J., Schiffmann, R., Brooks, B. P., Tamura, D., & DiGiovanna, J. J. (2007). Xeroderma pigmentosum, trichothiodystrophy, and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience, 145(4), 1388–1396. https://doi.org/10.1016/j.neuroscience.2006.08.039
- Patients should constantly supplement with vitamin D due to the stringent sun protection and avoidance.
- Patients can also obtain vitamin D from fortified foods, fish, eggs, mushrooms, and other vitamin D-rich foods.
- There is a contraindication of Radiation therapy in patients with XP. This is due to their DNA repair deficiency and high risk of tissue damage.10DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426
Complications of Disease:
Non-melanoma skin cancers are recurrent and develop in large numbers at the same time.
- The average age at which a person develops their first non-melanoma skin cancer is around nine.
- Invasive squamous cell carcinoma or metastatic malignant melanoma are the most frequent causes of mortality in XP patients. At the same time, neurodegeneration is the second most common factor that increases the risk of developing non-melanoma skin cancer.
- Premature Aging: Patients of xeroderma pigmentosa develop premature aging due to prolonged damage. Pathology in repairing skin damage depicts premature aging.
Life expectancy of Xeroderma Patients:
Depending on the specific XP subtype, people with xeroderma pigmentosum can expect a varying length of life. Comparatively, XP patients with neurologic signs often have shorter lifespans than those who do not have neurodegeneration.
- The most common causes of death in XP patients are metastatic malignant melanoma and invasive squamous cell carcinoma due to UV-induced skin cancers. Neurological decline is the second most common cause of death, particularly in XP subtypes such as XP-A and XP-D that are associated with central nervous system degeneration.
- For people with XP who do not have neurodegeneration, the average age at death is around 37.
- Patients with XP who have neurodegeneration die on average at a younger age, 29 years, than those without the condition.11DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426
Additionally, individuals with XP have a risk of more than 50 times higher for central nervous system tumors such as glioblastoma, medulloblastoma, and spinal cord astrocytoma. Smoking increases the incidence of lung cancer among XP patients compared to the general population.
Take-Home Message
Xeroderma pigmentosum is an autosomal recessive disorder that is impossible to treat completely with medications. Moreover, early diagnosis and prompt adaptability of life to manage the disease can increase the Quality of Life Index (QOLI) of patients. Emotional support for the patient is one of the important ways that a family can help him. Regular visits to a physician will decrease the probability of developing complications of high severity.
Refrences
- 1Black J. O. (2016). Xeroderma Pigmentosum. Head and neck pathology, 10(2), 139–144. https://doi.org/10.1007/s12105-016-0707-8
- 2Black J. O. (2016). Xeroderma Pigmentosum. Head and neck pathology, 10(2), 139–144. https://doi.org/10.1007/s12105-016-0707-8
- 3Lehmann, A. R., McGibbon, D., & Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet Journal of Rare Diseases, 6(70). https://doi.org/10.1186/1750-1172-6-70
- 4Lehmann, A. R., McGibbon, D., & Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet Journal of Rare Diseases, 6(70). https://doi.org/10.1186/1750-1172-6-70
- 5DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426
- 6Bradford, P. T., Goldstein, A. M., Tamura, D., Khan, S. G., Ueda, T., Boyle, J., … & Kraemer, K. H. (2011). Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. Journal of Medical Genetics, 48(3), 168–176. https://doi.org/10.1136/jmg.2010.082511
- 7Piccione, M., Belloni Fortina, A., Ferri, G., Andolina, G., Beretta, L., Cividini, A., De Marni, E., Caroppo, F., Citernesi, U., & Di Liddo, R. (2021). Xeroderma Pigmentosum: General Aspects and Management. Journal of personalized medicine, 11(11), 1146. https://doi.org/10.3390/jpm11111146
- 8Kleijer, W. J., Laugel, V., Berneburg, M., Nardo, T., Fawcett, H., Gratchev, A., … & Lehmann, A. R. (2008). Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair, 7(5), 744–750. https://doi.org/10.1016/j.dnarep.2008.01.004
- 9Kraemer, K. H., Patronas, N. J., Schiffmann, R., Brooks, B. P., Tamura, D., & DiGiovanna, J. J. (2007). Xeroderma pigmentosum, trichothiodystrophy, and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience, 145(4), 1388–1396. https://doi.org/10.1016/j.neuroscience.2006.08.039
- 10DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426
- 11DiGiovanna, J. J., & Kraemer, K. H. (2012). Shining a light on xeroderma pigmentosum. The Journal of Investigative Dermatology, 132(3), 785–796. https://doi.org/10.1038/jid.2011.426



